thalassemie
Tanja Unterberger studeerde journalistiek en communicatiewetenschap in Wenen. In 2015 begon ze haar werk als medisch redacteur bij in Oostenrijk. Naast het schrijven van specialistische teksten, tijdschriftartikelen en nieuws heeft de journalist ook ervaring met podcasting en videoproductie.
Meer over de experts Alle inhoud van wordt gecontroleerd door medische journalisten.Thalassemie, of mediterrane bloedarmoede, is een genetische ziekte van de rode bloedcellen. Een defect gen zorgt ervoor dat het lichaam te weinig rood bloedpigment (hemoglobine) aanmaakt of het wordt te snel afgebroken. Afhankelijk van de locatie van het genetisch defect wordt onderscheid gemaakt tussen alfa- en bèta-thalassemie. Beide vormen leiden tot bloedarmoede (bloedarmoede). De arts behandelt deze meestal met medicijnen en bloedtransfusies. Lees hier meer over oorzaken, symptomen, diagnose en behandeling!
ICD-codes voor deze ziekte: ICD-codes zijn internationaal erkende codes voor medische diagnoses. Ze staan bijvoorbeeld in doktersbrieven of op attesten van arbeidsongeschiktheid. D57D56

Kort overzicht
- Beschrijving: Genetisch bepaalde ziekte van de rode bloedcellen (erytrocyten) die leidt tot bloedarmoede.
- Diagnose: De arts stelt thalassemie vast door middel van een speciaal bloedonderzoek en een analyse van het genetisch materiaal (DNA-analyse).
- Oorzaken: Een erfelijke genetische afwijking waardoor het lichaam te weinig of geen rood bloedpigment (hemoglobine) aanmaakt.
- Behandeling: De behandeling bestaat uit medicatie en bloedtransfusies, in sommige gevallen is een beenmergtransplantatie noodzakelijk.
- Symptomen: Onder andere bloedarmoede, vermoeidheid, een vergrote lever en milt, groeistoornissen, botveranderingen, osteoporose; milde vormen van thalassemie hebben vaak geen symptomen.
- Prognose: thalassemieën hebben verschillende gradaties van ernst. Hoe eerder de symptomen worden behandeld, hoe beter de prognose. Genezing is momenteel alleen mogelijk met een stamceltransplantatie of gentherapie.
Wat is thalassemie?
Thalassemieën (ook mediterrane anemie) zijn een groep genetische ziekten waarbij de vorming van rode bloedcellen (erytrocyten) wordt verstoord door een genetisch defect. Hierdoor maakt het lichaam te weinig of geen rood bloedpigment (hemoglobine) aan of wordt het te snel afgebroken.
Als gevolg van de veranderde hemoglobine zijn de bloedcellen meestal kleiner dan normaal en hebben ze een kortere levensduur, wat na verloop van tijd leidt tot bloedarmoede, gepaard gaand met typische symptomen zoals vermoeidheid en versnelde hartslag.
Thalassemie is een van de meest voorkomende bloedziekten bij kinderen en adolescenten. Ze behoren ook tot de groep van hemoglobinopathieën (aandoeningen van hemoglobine). Thalassemie kan van generatie op generatie worden doorgegeven. Aangezien thalassemie bijzonder wijdverbreid is in het Middellandse Zeegebied, wordt het ook wel "mediterrane bloedarmoede" genoemd.
Hoe ontwikkelt thalassemie zich?
Hemoglobine is een eiwit dat voorkomt in rode bloedcellen en wordt gemaakt in het beenmerg. Het stelt de rode bloedcellen in staat om vitale zuurstof van de longen naar alle delen van het lichaam te transporteren.
Hemoglobine bestaat meestal uit vier eiwitketens, waarvan er twee hetzelfde zijn: twee alfa- en twee bètaketens. Hemoglobine bevat ook ijzer, dat zuurstof bindt. Bij thalassemie vormt het lichaam geen, te weinig of veranderde eiwitketens (alfa- of bètaketens) door een veranderd gen (mutatie).
Dit betekent dat er zich minder functionele hemoglobinemoleculen kunnen vormen. De rode bloedcellen krimpen en breken steeds meer af. Dit leidt tot bloedarmoede, omdat het lichaam door een tekort aan rode bloedcellen niet meer voldoende van zuurstof wordt voorzien.
Afhankelijk van welke eiwitketen (alfa- of bètaketen) is aangetast, wordt onderscheid gemaakt tussen α- (alfa)-thalassemie en β- (beta)-thalassemie.
Alfa-thalassemie
De minder voorkomende vorm is α-thalassemie. In deze vorm vormt het lichaam te weinig of geen alfaketens.
Bèta-thalassemie
Bèta-thalassemie is de meest voorkomende vorm van thalassemie. In deze vorm maakt het lichaam te weinig of geen bèta-hemoglobineketens aan.
Er is ook delta- en gamma-thalassemie, maar beide zijn zeer zeldzaam en meestal mild.
Hoe wordt thalassemie gediagnosticeerd?
Aangezien de symptomen van thalassemie vaak in de kindertijd verschijnen, is het eerste aanspreekpunt meestal de kinderarts. Indien nodig en voor nader onderzoek verwijst hij u door naar een specialist interne geneeskunde (hematoloog).
Familiegeschiedenis
Aangezien thalassemie een erfelijke ziekte is, geeft de familiegeschiedenis de arts meestal de eerste aanwijzingen. De arts vraagt bijvoorbeeld of er in de familie dragers van de ziekte zijn.
Bloed Test
Om de diagnose te bevestigen, zal de arts een bloedtest uitvoeren, inclusief hemoglobine-elektroforese (Hb-elektroforese).
Hemoglobine-elektroforese
Bij hemoglobine-elektroforese lost de arts hemoglobine uit het bloed van de patiënt op in een vloeistof en brengt dit aan op een speciaal dragermateriaal (bijvoorbeeld van papier of gelatine). Dan zet hij een spanning op.
Afhankelijk van hoe de hemoglobine is samengesteld, legt het in een bepaalde tijd verschillende afstanden af. Op basis van de afgelegde afstand beoordeelt de arts of de hemoglobine normaal of defect is.
Onder een microscoop herkent de arts meestal de karakteristiek gevormde cellen in het bloed van de persoon. Dit zijn relatief bleke rode bloedcellen met een donkerder gekleurde afzetting van hemoglobine in het midden. De bloedcellen zijn klein en hebben een slechte hemoglobine.
Diagnose bij baby's
Artsen raden aan dat pasgeborenen die thalassemie in de familie hebben gehad, een bloedtest ondergaan (bijvoorbeeld voor screening van pasgeborenen). Hiervoor neemt de arts direct na de geboorte wat bloed (bijvoorbeeld uit de navelstreng) en onderzoekt dit op genetische veranderingen. Op deze manier is het mogelijk om de ziekte vroegtijdig te herkennen en tijdig te behandelen.
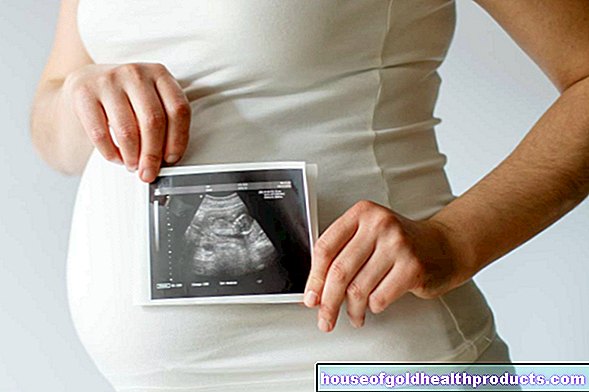
Diagnose tijdens de zwangerschap
Bij ongeboren kinderen wordt het onderzoek naar thalassemie (prenatale diagnose) uitgevoerd met behulp van een weefselmonster uit de placenta.
Artsen raden aanstaande ouders die weten dat ze drager zijn van thalassemie aan om deze prenatale diagnose in de eerste twaalf weken van de zwangerschap te laten stellen.
Is thalassemie erfelijk?
Thalassemie is erfelijk. Dit betekent dat mensen met thalassemie het genetische defect van hun ouders erven en met de ziekte worden geboren.
Overerving: alfa-thalassemie
Alfa-thalassemie heeft verschillende gradaties van ernst. Er wordt onderscheid gemaakt tussen alfa-thalassemie minima, minor, intermedia en major. De expressie hangt af van het aantal pathologische genen dat ouders doorgeven aan het kind. Er zijn vier genen voor de alfaketen, twee van de moeder en twee van de vader.
In totaal zijn vier genen betrokken bij de vorming van de alfaketens. Bij alfa-thalassemie major zijn alle genen die het kind van de ouders heeft geërfd defect. Het is de meest ernstige vorm van alfa-thalassemie. Kinderen met deze vorm zijn zelden in staat om te overleven en sterven meestal voor de geboorte of enkele dagen daarna. Slechts in enkele gevallen is het mogelijk om de kinderen in leven te houden met behulp van een stamceltransplantatie.
Mensen met alfa-thalassemie minor of intermedia erfden twee of drie defecte genen van hun ouders. Ze behoren tot de mildere vormen van alfa-thalassemie en veroorzaken meestal matige, milde of geen symptomen bij de getroffenen.
Als slechts één ouder een defect gen van hun kind erft, wordt dit alfa-thalassemie minima genoemd. De getroffenen hebben meestal geen symptomen en leven zonder beperkingen.
Overerving: bèta-thalassemie
Bèta-thalassemie heeft ook verschillende gradaties van ernst. Ze zijn onderverdeeld in minor, intermedia en major.
Thalassemie major (ook wel Cooley's anemie genoemd) is de meest ernstige vorm. Het treedt op wanneer beide ouders het genetisch defect doorgeven aan het kind.
Bij bèta-thalassemie major is het noodzakelijk om het bloed van de aangedane persoon regelmatig (ongeveer elke derde tot vierde week) te verversen door middel van infusies. Indien onbehandeld, ontwikkelen de getroffenen meestal ernstige botvervormingen. De getroffen kinderen zijn ook veel vatbaarder voor infecties, ze zijn zwak en ontwikkelen zich meestal niet volgens hun leeftijd.
Beta-thalassemie intermedia is een tussenvorm. Mensen met deze vorm erfden ook de defecte genen van beide ouders. Hun symptomen zijn meestal niet zo uitgesproken als in de hoofdvorm. Bloedtransfusies heb je maar af en toe nodig.
Bij bèta-thalassemie minor hebben de getroffenen slechts één defect bèta-hemoglobineketengen van één ouder geërfd. De andere ouder heeft een werkend gen doorgegeven aan het kind.
Mensen met bèta-thalassemie minor hebben meestal geen tot milde symptomen van bloedarmoede. In de regel hebben ze geen behandeling nodig. Als gendragers kunnen ze de thalassemie echter doorgeven aan hun kinderen.
Als beide ouders kleine bèta-thalassemie hebben, is er een kans van 25 procent dat het kind beide mutaties zal erven en grote of intermediale thalassemie zal ontwikkelen. Er is 50 procent kans dat het kind een minderjarige vorm erft en 25 procent kans dat het kind gezond is.
Om bèta-thalassemie te krijgen, moeten beide ouders hun defecte genen doorgeven aan het kind. Een drager van de ziekte heeft slechts één defect gen, het andere gen is functioneel. Hoewel ze niet ziek worden, kunnen ze het gen wel doorgeven aan hun kinderen.
Waar komt thalassemie voor?
Thalassemie is ook in de volksmond bekend als mediterrane bloedarmoede, omdat het wijdverbreid is in mediterrane landen zoals Italië en Griekenland. Maar het komt ook voor in het Nabije en Midden-Oosten en in delen van Afrika en Azië.In Centraal-Europa heeft thalassemie zich verspreid via wereldwijde handelsbetrekkingen en migratie.
Sinds de jaren zestig komt thalassemie ook voor in Centraal- en Noord-Europa (o.a. Duitsland, Oostenrijk, Frankrijk, Engeland, Nederland, België en Scandinavië). Ondertussen is thalassemie een van de meest voorkomende bloedziekten bij kinderen en adolescenten.
Hoe vaak komt de ziekte voor?
Naar schatting leven in Duitsland zo'n 500 tot 600 mensen met een ernstige vorm van thalassemie. Ongeveer 200.000 mensen in Duitsland lijden aan de milde vorm van thalassemie. Bovendien komen bèta-thalassemieën vaker voor dan alfa-thalassemieën.
Hoe behandel je thalassemie?
De arts, meestal een specialist (bijvoorbeeld een kinderhematoloog), behandelt thalassemie afhankelijk van de ernst van de ziekte en de symptomen die optreden. De behandeling vindt plaats in centra die gespecialiseerd zijn in de ziekte. Afhankelijk van of de getroffen persoon een bloedtransfusie nodig heeft, wordt nu onderscheid gemaakt tussen transfusieafhankelijke (TDT) en niet-transfusieafhankelijke (NTDT) therapie voor thalassemie.
Niet-transfusieafhankelijke behandeling
Milde vormen van thalassemie vereisen meestal geen behandeling. In zeldzame gevallen tijdens de zwangerschap kunnen zwangere vrouwen echter ernstige bloedarmoede krijgen als gevolg van milde thalassemie. Ze hebben dan regelmatig bloedtransfusies nodig om zowel moeder als kind te beschermen.
Transfusieafhankelijke behandeling
De arts wisselt het bloed van transfusieafhankelijke mensen (bijvoorbeeld met thalassemie major of intermedia) uit met regelmatige bloedtransfusies. Afhankelijk van de ernst van de thalassemie vindt dit meestal ongeveer elke tweede tot derde week plaats, meestal een heel leven lang.
De extra bloedtransfusies leiden vaak tot een teveel aan ijzer. In deze gevallen zal de arts medicijnen voorschrijven die het overtollige ijzer uit het lichaam verwijderen (zogenaamde chelatoren of ijzerchelatoren). De arts injecteert deze onderhuids (subcutaan) in het ziekenhuis of de betrokkene neemt thuis tabletten in.
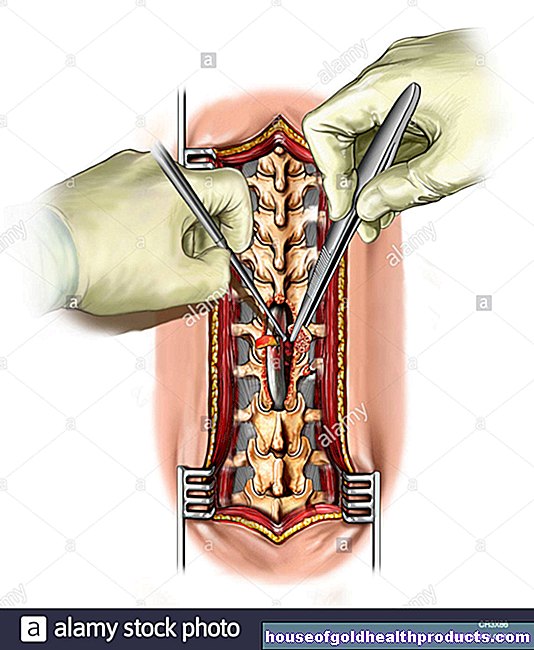
Stamceltransplantatie
Het is mogelijk om thalassemie major te genezen met een stamceltransplantatie. Dit veronderstelt echter dat er een geschikte donor is voor de betrokkene.
Gentherapie
Sinds 2019 is er gentherapie voor transfusieafhankelijke bèta-thalassemie. De therapie behandelt het causale genetische defect en geeft de getroffenen de kans om levenslang zonder transfusies te leven.
Bij deze nieuwe behandeling wordt een intact gen dat in staat is om gezond hemoglobine te produceren, in de bloedstamcellen van de persoon geïnjecteerd. Hiervoor worden eerst stamcellen uit het beenmerg van de patiënt gehaald en behandeld met een medicijn. Dan krijgt betrokkene zijn eigen stamcellen (met intacte genen) terug, die nu gezonde rode bloedcellen en normaal bloedpigment vormen.
Het Europees Geneesmiddelenbureau (EMA) heeft de behandeling goedgekeurd voor mensen van twaalf jaar en ouder die geschikt zijn voor deze specifieke vorm van therapie. Gentherapie is momenteel alleen mogelijk in Duitsland. Momenteel worden er nog maar weinig patiënten mee behandeld.
Dieet en levensstijl
Mensen met thalassemie kunnen het verloop van de ziekte positief beïnvloeden door de juiste voeding te eten en een passende levensstijl aan te nemen. Let op het volgende:
- Eet een gebalanceerd dieet.
- Vermijd voedingsmiddelen die extreem veel ijzer bevatten (bijv. lever).
- Haal voldoende calcium uit voedsel (bijv. melk, yoghurt, kaas, spinaziebladeren, broccoli).
- Drink geen alcohol.
- Zorg voor regelmatige lichaamsbeweging (ongeveer drie tot vier uur lichaamsbeweging, zoals snel wandelen, joggen of fietsen per week).
Wat zijn de symptomen van thalassemie?
Hoe ernstig de symptomen zijn, hangt af van hoeveel van de hemoglobine abnormaal is veranderd. Mensen met een milde vorm van thalassemie hebben meestal weinig of geen symptomen.
Indien onbehandeld, vertonen baby's met een ernstige vorm van thalassemie echter al op de leeftijd van vier of vijf maanden gezondheidsproblemen.
Bloedarmoede
Mensen met thalassemie major of intermedia vertonen aanvankelijk typische symptomen van bloedarmoede:
- Ze zijn bleek of hebben een gelige huid.
- Je bent de hele tijd moe.
- Je voelt je duizelig.
- Jij hebt hoofdpijn.
- Je hebt een snelle hartslag.
- Ze worden snel kortademig tijdens lichamelijke activiteit.
- Ze hebben vaak een vergrote lever en milt.
- Hun ontwikkeling is vaak verstoord.
Op het eerste gezicht lijkt thalassemie op bloedarmoede door ijzertekort. Het ijzergehalte is echter laag bij ijzertekort, terwijl het bij thalassemie normaal is.
Later of als de behandeling niet adequaat is, ervaren de getroffenen vaak ernstige bijwerkingen. Deze omvatten bijvoorbeeld:
- Botvervormingen
- Frequente infecties
- hart problemen
- Suikerziekte
- osteoporose
Bot veranderingen
Mensen met thalassemie major of intermedia vertonen vaak ook abnormale botgroei:
- Het schedelbeen wordt groter.
- Het voorhoofd, de bovenkaak en het jukbeen zijn uitpuilend.
- Ribben en wervellichamen zijn gebogen.
- U heeft een verhoogd risico op fracturen.
- Je hebt botpijn.
- Je krijgt osteoporose.
Overtollig ijzer
Mensen met ernstige vormen van thalassemie slaan vaak te veel ijzer op in het lichaam (ijzerstapeling), dat niet meer vanzelf afbreekt. Dit kan de organen beschadigen (secundaire hemochromatose). Het verhoogde ijzer komt onder meer van het extra bloed dat de getroffen persoon regelmatig via infusies krijgt.
Daarnaast beschouwt het lichaam de bloedarmoede als een ijzertekort en probeert het dit te compenseren door meer ijzer uit de voeding op te nemen. Het lichaam maakt ook overmatige bloedcellen aan. De lever en milt werken dan op volle toeren om ze weer af te breken.
De te hoge ijzerconcentratie veroorzaakt andere symptomen bij de getroffenen, zoals:
- Hartfalen (hartfalen)
- Hartritmestoornissen
- lever disfunctie
- Diabetes (diabetes mellitus)
- Korte gestalte
- Vertraagde puberteit
- Traag werkende schildklier (hypothyreoïdie)
- Aandoeningen van het vitamine D-metabolisme
Wanneer naar de dokter?
Aangezien ernstige vormen van thalassemie vaak in de eerste levensmaanden optreden, is het belangrijk dat ouders bij de eerste tekenen (bijvoorbeeld bleekheid) zo snel mogelijk een kinderarts zien.
Hoe kun je het voorkomen?
Artsen adviseren mensen die weten dat ze het gen voor thalassemie hebben om voor een geplande zwangerschap of als ze kinderen willen krijgen naar een genetisch adviescentrum te gaan.
Getrainde specialisten gebruiken een genetische bloedtest om de mogelijke genetische risico's van zwangerschap bij paren te bepalen.
Mensen met een bekende familiegeschiedenis van thalassemie wordt ook geadviseerd om advies in te winnen voordat ze zwanger worden.
Om levensbedreigende infecties te voorkomen, adviseren artsen ook om kinderen met thalassemie en gezonde kinderen te vaccineren volgens de huidige vaccinatiekalender. In sommige gevallen is het nodig om kinderen met thalassemie regelmatig te behandelen met bepaalde antibiotica. Deze helpen ernstige bacteriële infecties te voorkomen, waar mensen met thalassemie bijzonder vatbaar voor zijn.
Regelmatige meting van de lichaamstemperatuur helpt om infecties zo vroeg mogelijk op te sporen en zo snel mogelijk te behandelen. Als u koorts heeft van meer dan 38,5 graden Celsius, moet u onmiddellijk een arts raadplegen. Het is mogelijk dat een infectie dit veroorzaakt.
Het is ook bijzonder belangrijk dat de getroffenen zich regelmatig door een arts laten controleren. Alleen zo kan het ziekteverloop goed worden gevolgd en complicaties in een vroeg stadium worden gesignaleerd en behandeld.
Is thalassemie te genezen?
Een volledige genezing van thalassemie is momenteel alleen mogelijk met een stamceltransplantatie of met gentherapie. De levensverwachting en kwaliteit van de getroffenen is voortdurend gestegen dankzij nieuwe medicijnen, verbeterde behandeling en beter onderwijs.
Wat is de prognose voor thalassemie?
Mensen met milde thalassemie (kleine alfa- of bèta-thalassemie) hebben een normale levensverwachting. In de regel is het voor de getroffenen mogelijk om zonder beperkingen te leven.
Bij ernstigere vormen is het vaak ook mogelijk om met de juiste en vroege behandeling (bijvoorbeeld regelmatige bloedtransfusies) een bijna normale levensverwachting te bereiken. Wel is dan een levenslange, continue en intensieve therapie noodzakelijk.
Soms kan stamceltransplantatie de ziekte genezen en, in zeldzame gevallen, gentherapie.
Tags: laboratoriumwaarden sekspartnerschap digitale gezondheid