Sikkelcelanemie
Tanja Unterberger studeerde journalistiek en communicatiewetenschap in Wenen. In 2015 begon ze haar werk als medisch redacteur bij in Oostenrijk. Naast het schrijven van specialistische teksten, tijdschriftartikelen en nieuws heeft de journalist ook ervaring met podcasting en videoproductie.
Meer over de experts Alle inhoud van wordt gecontroleerd door medische journalisten.Sikkelcelziekte (ook wel sikkelcelziekte of drepanocytose) is een erfelijke ziekte waarbij de rode bloedcellen sikkelvormig worden. De oorzaak is een genetisch defect. Symptomen zijn onder meer stoornissen in de bloedsomloop en bloedarmoede. Genezing is momenteel alleen mogelijk met een stamceltransplantatie. Lees hier meer over oorzaken, symptomen en therapie!
ICD-codes voor deze ziekte: ICD-codes zijn internationaal erkende codes voor medische diagnoses. Ze staan bijvoorbeeld in doktersbrieven of op attesten van arbeidsongeschiktheid. D57

Kort overzicht
- Beschrijving: Erfelijke ziekte waarbij de rode bloedcellen (erytrocyten) sikkelvormig worden
- Oorzaken: De oorzaak van sikkelcelanemie is een defect gen dat verantwoordelijk is voor de vorming van hemoglobine (rood bloedpigment).
- Prognose: Sikkelcelanemie heeft een variërende mate van ernst. Hoe eerder de symptomen worden behandeld, hoe beter de prognose. Indien onbehandeld, is de ziekte meestal dodelijk.
- Symptomen: Ernstige pijn, stoornissen in de bloedsomloop, bloedarmoede, frequente infecties, orgaanschade (bijv. milt), beroertes, groeistoornissen
- Behandeling: De symptomen van sikkelcelziekte kunnen worden verlicht met medicijnen (bijv. hydroxycarbamide, pijnstillers) en bloedtransfusies. Genezen is mogelijk met een stamceltransplantatie.
- Diagnose: gesprek met de arts, lichamelijk onderzoek, bloedonderzoek, echografie, CT, MRI
Wat is sikkelcelanemie?
Sikkelcelziekte - ook bekend als sikkelcelziekte (SCD) of drepanocytose - is een erfelijke ziekte. Tijdens de ziekte veranderen gezonde rode bloedcellen (erytrocyten) in abnormale, sikkelvormige cellen (sikkelcellen). Door hun vorm kunnen deze bloedvaten in het lichaam verstoppen. Typische symptomen van de ziekte zijn hevige pijn, stoornissen in de bloedsomloop, bloedarmoede en orgaanschade.
Sikkelcelanemie wordt veroorzaakt door een veranderd gen dat verantwoordelijk is voor de vorming van het rode bloedpigment (hemoglobine). Sikkelcelziekte is niet besmettelijk en bestaat al bij de geboorte. De eerste symptomen verschijnen vaak in de kindertijd of peutertijd.
De ziekte behoort tot de groep van hemoglobinopathieën. Dit zijn verschillende aandoeningen van het rode bloedpigment hemoglobine.
Artsen geven de voorkeur aan de term sikkelcelziekte in plaats van sikkelcelanemie, omdat niet alle vormen geassocieerd zijn met bloedarmoede. Bovendien is de bloedarmoede niet het belangrijkste symptoom van de ziekte, maar de symptomen die de vasculaire occlusie veroorzaken.
Hoe vaak komt sikkelcelanemie voor?
Sikkelcelanemie is een van de meest voorkomende erfelijke ziekten en de meest voorkomende bloedziekte ter wereld. Er zijn momenteel ongeveer 3.000 tot 5.000 mensen in Duitsland met sikkelcelziekte. Volgens een schatting van de Wereldgezondheidsorganisatie (WHO) worden jaarlijks wereldwijd meer dan 300.000 kinderen geboren met sikkelcelziekte.
Wie wordt in het bijzonder getroffen?
Sikkelcelanemie treft vooral mensen uit Centraal- en West-Afrika. Het kwam aanvankelijk vooral voor in het sub-Sahara deel van het Afrikaanse continent. Maar door migratie is sikkelcelziekte inmiddels wereldwijd wijdverbreid. Tegenwoordig worden ook veel mensen uit delen van de Middellandse Zee, het Midden-Oosten, India en Noord-Amerika getroffen.
Sikkelcelziekte is sinds de jaren zestig ook wijdverbreid in Noord-Europa (bijv. Duitsland, Oostenrijk, Frankrijk, Engeland, Nederland, België en Scandinavië).
Sikkelcelanemie en malaria
Mensen met sikkelcelziekte zijn minder vatbaar voor malaria. De reden hiervoor: De rode bloedcellen transporteren normaal gesproken de malariapathogenen (plasmodia) via het bloed door het lichaam. Bij sikkelcelziekte veranderen de rode bloedcellen in sikkelcellen en zijn ze minder mobiel. De malariapathogenen overleven daardoor slechter.
Dit geeft mensen met een gezond en een ziek gen (zogenaamde heterozygote gendragers) een overlevingsvoordeel (heterozygoot voordeel) in de regio's waar malaria zich verspreidt. Dit is ook de verklaring waarom relatief veel mensen daar ziek zijn van sikkelcelanemie.
Hoe ontstaat sikkelcelanemie?
Sikkelcelanemie is een erfelijke ziekte. De oorzaak is een veranderd gen (mutatie) dat de vader en moeder doorgeven aan hun kind. Het genetische defect verandert gezond, rood bloedpigment (hemoglobine; kortweg Hb) in sikkelcelhemoglobine (hemoglobine S; kortweg HbS).
Aangeboren genetisch defect
De rode bloedcellen hebben de vitale taak om zuurstof van de longen naar alle delen van het lichaam te transporteren. Om dit te doen, bindt de zuurstof zich aan het rode bloedpigment. Dit is een eiwit dat voorkomt in rode bloedcellen.
De rode bloedcellen bestaan normaal gesproken uit "gezond" hemoglobine, dat is opgebouwd uit twee eiwitketens - de alfa- en bètaketens. Deze ketens maken de bloedcellen rond en glad, waardoor ze door elk klein bloedvat passen en alle organen van vitale zuurstof en voedingsstoffen voorzien.
Bij sikkelcelanemie betekenen genetische fouten (mutaties) dat de zogenaamde bètaketen van hemoglobine abnormaal verandert (hemoglobine S).Als er te weinig zuurstof in het bloed zit, verandert de vorm van de sikkelcelhemoglobine en daarmee die van de rode bloedcellen.
De hemoglobinedeeltjes vormen starre vormen en klonteren samen, waardoor de rode bloedcellen de typische vorm aannemen en eruitzien als kleine sikkels. Dit is waar de term "sikkelcelziekte" vandaan komt.
Door hun vorm zijn sikkelcellen veel onbeweeglijker en sterven ze eerder af dan gezonde rode bloedcellen (hemolyse). Dit resulteert enerzijds in bloedarmoede in het lichaam (zogenaamde corpusculaire hemolytische anemie) omdat de betrokkene te weinig rode bloedcellen heeft. Anderzijds verstoppen de sikkelcellen door hun vorm gemakkelijker kleinere bloedvaten.
Dit betekent dat de aangetaste delen van het lichaam niet langer voldoende van zuurstof worden voorzien. Hierdoor kan het weefsel afsterven en in het ergste geval kunnen de organen hun vitale functies niet meer vervullen.
Hoe wordt sikkelcelziekte geërfd?
Sikkelcelanemie wordt geërfd van zowel vader als moeder. Het is een zogenaamde autosomaal recessieve overerving: elke ouder draagt minstens één pathologisch gen in de chromosomen (drager van de genetische informatie).
Het heeft verschillende effecten of het kind slechts één pathologisch gen of beide genen erft:
Homozygote gendrager
Als zowel de vader als de moeder het gemodificeerde gen in zich dragen en ze geven het allebei door aan hun kind, dan draagt het kind twee gemodificeerde genen (homozygote gendrager). Het lijdt aan sikkelcelanemie. In dit geval maakt het kind alleen pathologisch hemoglobine (HbS) aan en geen gezond hemoglobine (Hb).
Heterozygote gendrager
Als de ene ouder een gezond gen doorgeeft en de andere ouder een ziek gen, maakt het kind zowel gezond hemoglobine als sikkelcelhemoglobine aan. In dit geval krijgt het kind geen sikkelcelanemie. Het draagt echter het pathologische gen en kan het later doorgeven aan zijn kinderen (heterozygote gendrager).
Artsen adviseren dragers van de ziekte ook om advies in te winnen over de mogelijke erfenis en de bijbehorende risico's voor het kind als ze kinderen willen krijgen
Is sikkelcelziekte te genezen?
De prognose van sikkelcelanemie hangt sterk af van hoe goed en vroeg de symptomen en complicaties worden behandeld. Ongeveer 85 tot 95 procent van alle kinderen met sikkelcelziekte bereikt de volwassen leeftijd in landen met goede gezondheidszorg (bijv. Europa, VS). De gemiddelde levensverwachting ligt dan rond de 40 tot 50 jaar. In landen met een slechtere medische zorg is de mortaliteit hoger.
Met bloedstamceltransplantaties is het nu mogelijk voor artsen om mensen met sikkelcelanemie volledig te genezen. Deze behandeling brengt echter ook enkele risico's met zich mee en is daarom voorbehouden aan ernstige gevallen van sikkelcelanemie.
Hoe gaat het met de ziekte?
Sommige mensen met sikkelcelziekte hebben weinig symptomen, terwijl anderen veel last hebben van de gevolgen van de ziekte. Het is nog niet duidelijk waarom de ziekte zo anders verloopt.
Mensen met sikkelcelanemie vertonen meestal de eerste tekenen van de ziekte als baby.
Welke soorten sikkelcelziekte zijn er?
Er zijn verschillende vormen van sikkelcelanemie. Deze ontstaan afhankelijk van hoe het gen dat verantwoordelijk is voor de vorming van het rode bloedpigment wordt veranderd. Alle vormen zijn erfelijk. De respectievelijke vormen lopen echter anders en de symptomen zijn ook anders uitgesproken.
De meest voorkomende vormen van sikkelcelziekte zijn:
Sikkelcelziekte HbSS (SCD-S/S)
De SCD-S/S is de meest voorkomende vorm van sikkelcelanemie. Het is het moeilijkst. De getroffenen hebben elk een gemodificeerd gen geërfd van beide ouders. Daarom vormen ze alleen het pathologisch veranderde sikkelcelhemoglobine (HbS).
Sikkelcelziekte HbSß-Thal (SCS-S / beta-Thal)
Met SCS-S/bèta-Thal erven kinderen het sikkelcelgen van de ene ouder en een gen voor de zogenaamde bèta-thalassemie van de andere. Dit laatste is een andere bloedziekte waarbij het lichaam te weinig of geen hemoglobine aanmaakt. Deze vorm van sikkelcelziekte is zeldzamer en meestal milder.
Sikkelcelziekte HbSC (SCD-S/C)
Bij SCD-S/C erft één ouder het gen voor sikkelcelziekte en de andere ouder erft een veranderd hemoglobinegen, het HbC-gen. Hoge bloedspiegels van het rode bloedpigment hemoglobine zijn typisch. Omdat het bloed daardoor dikker is, kunnen complicaties optreden, zoals stoornissen in de bloedsomloop. Deze vorm van sikkelcelanemie is meestal minder ernstig, maar kan in sommige gevallen leiden tot blindheid of doofheid.
Sikkelcelziekte ontstaat niet alleen wanneer een kind twee sikkelcelgenen erft, maar ook wanneer de ouders een sikkelcel-gen doorgeven in combinatie met een ander pathologisch veranderd hemoglobine-gen (zoals het HbC- of thalassemie-gen).
Wat zijn de symptomen?
De symptomen die optreden bij sikkelcelanemie treffen meestal het hele lichaam. In principe is het mogelijk dat de sikkelcellen elk bloedvat in het lichaam verstoppen. De meest voorkomende symptomen van sikkelcelziekte zijn:
Sterke pijn
Mensen met sikkelcelziekte hebben meestal hevige pijn (zogenaamde pijncrises). Je lichaam maakt stoffen aan die pijn veroorzaken wanneer de sikkelcellen de bloedvaten verstoppen en daardoor de bloedtoevoer naar de organen (vooral het beenmerg) en weefsels blokkeren. De belangrijkste pijn in uw maag, botten en gewrichten is tijdens een crisis.
Weersveranderingen, gebrek aan vocht, infecties met koorts en uitputting veroorzaken vaak deze pijn. Ouderen met sikkelcelanemie worden ook vaker getroffen door ernstige pijncrises dan kinderen.
Sommige mensen met sikkelcelziekte hebben kortdurende pijn, terwijl anderen vaak lange tijd in het ziekenhuis doorbrengen en pijnstillers nodig hebben.
Bloedarmoede
Sikkelcellen zijn minder stabiel dan gezonde rode bloedcellen en sterven daardoor eerder af (hemolyse). Sikkelcellen overleven ongeveer tien tot twintig dagen, terwijl rode bloedcellen meestal na ongeveer 120 dagen worden afgebroken. Als er te weinig rode bloedcellen in het lichaam zijn, ontstaat er na verloop van tijd bloedarmoede en neemt de concentratie van het rode bloedpigment af.
orgaanschade
Doordat de bloedvaten door de sikkelcellen worden geblokkeerd, worden de aangetaste delen van het lichaam niet meer voldoende van bloed voorzien (zogenaamde sikkelcelcrises). Als gevolg hiervan krijgt het weefsel te weinig zuurstof en voedingsstoffen, wat op den duur tot de dood leidt. De aangetaste organen kunnen dan niet meer goed functioneren.
Dit treft vooral het beenmerg, de longen, de hersenen, de milt en het maag-darmkanaal.
infecties
Als sikkelcelanemie de milt in de eerste levensjaren aantast, lopen patiënten een hoog risico op ernstige infecties. De reden hiervoor is dat de milt een belangrijke rol speelt bij het bestrijden van ziektekiemen (bijvoorbeeld bacteriën zoals pneumokokken en meningokokken, die longontsteking of meningitis veroorzaken) in het lichaam.
Infecties met koorts zijn een noodgeval bij mensen met sikkelcelziekte! Raadpleeg daarom direct een arts als de lichaamstemperatuur hoger is dan 38,5 graden Celsius!
Hand-voet syndroom
Door de vaatafsluitingen ervaren kleine kinderen vaak pijn, roodheid en zwelling van de handen en voeten (hand-voetsyndroom). Dit is vaak het eerste teken dat het kind sikkelcelziekte heeft.
groeiachterstand
Door de vaatafsluitingen ervaren kleine kinderen vaak pijn, roodheid en zwelling van de handen en voeten (hand-voetsyndroom). Dit is vaak het eerste teken dat het kind sikkelcelziekte heeft.
Botnecrose
Doordat de sikkelcellen de bloedvaten verstoppen, sterft ook weefsel in gewrichten of botten na verloop van tijd af (avasculaire botnecrose). De heupgewrichten en schoudergewrichten zijn vaak aangetast.
zweren
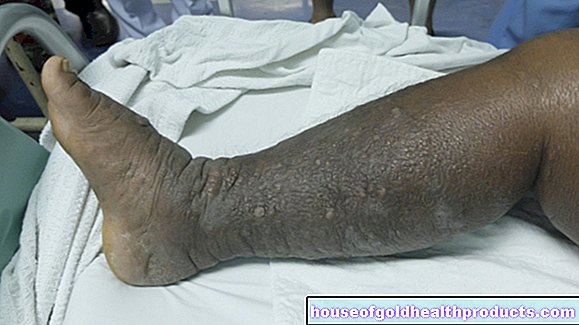
Ook is het mogelijk dat sikkelcellen de bloedvaten in de huid (vooral op de benen) verstoppen, waardoor het weefsel niet meer voldoende van voedingsstoffen wordt voorzien. Als gevolg hiervan hebben de getroffenen vaak pijnlijke, open wonden op hun benen (ulcera) die meestal moeilijk te genezen zijn.
Visuele stoornissen en blindheid
Wanneer de sikkelcellen de bloedvaten in het netvlies van de ogen verstoppen, sterft het omringende weefsel af en vormen zich littekens aan de achterkant van het oog. Dit beïnvloedt het gezichtsvermogen van de getroffenen en kan in het ergste geval leiden tot blindheid.
Galstenen, geelzucht
Doordat de sikkelvormige rode bloedcellen sneller desintegreren bij sikkelcelanemie, ontstaan er meer afbraakproducten in het bloed, bijvoorbeeld bilirubine (galpigment). Bilirubine wordt geproduceerd wanneer het rode bloedpigment hemoglobine wordt afgebroken. Soms vormen deze afbraakproducten galstenen in de galblaas. Als deze vast komen te zitten in de galwegen, kan dit leiden tot hevige pijn (galkoliek) en geelzucht (geelzucht). Galblaasontsteking is ook mogelijk als complicatie van sikkelcelziekte.
Hemolytische crisis, aplastische crisis
Bij sikkelcelziekte worden de sikkelvormige rode bloedcellen sneller afgebroken. Er bestaat een risico op hemolytische crises, bijvoorbeeld veroorzaakt door infecties, waarbij enorme hoeveelheden rode bloedcellen worden afgebroken. Als er helemaal geen rode bloedcellen worden aangemaakt, spreken artsen van een aplastische crisis. De getroffenen hebben onmiddellijke medische behandeling nodig! Vaak is een bloedtransfusie nodig om levensbedreigend zuurstoftekort en hart- en vaatziekten te voorkomen.
Miltsekwestratie en miltvergroting
De milt is het orgaan waarin de rode bloedcellen worden afgebroken. Bij mensen met sikkelcelanemie is het mogelijk dat grote hoeveelheden bloed plotseling in de milt "zakken" (miltsekwestratie). Dit leidt soms tot levensbedreigende bloedarmoede en zuurstofgebrek.
Miltsekwestratie ontwikkelt zich gewoonlijk gedurende één tot drie dagen. Symptomen zijn onder meer koorts en buikpijn. De getroffenen zijn vaak bleek en slap, vergelijkbaar met een verkoudheid. De milt zwelt op door de hoeveelheid bloed die zakt. In de meeste gevallen is dit voelbaar.
Het is belangrijk dat ouders van baby's en jonge kinderen met sikkelcelziekte de milt van hun kind leren palperen. Als de milt is vergroot, adviseren artsen ouders om hun kinderen onmiddellijk naar het ziekenhuis te brengen.
Acuut thoracaal syndroom (ATS)
Wanneer sikkelcellen bloedvaten in de longen verstoppen, kan het zogenaamde acuut thoracaal syndroom (kortweg ATS) optreden. Naast miltsequestratie is acuut thoracaal syndroom een van de meest voorkomende doodsoorzaken bij mensen met sikkelcelziekte. Een acuut borstsyndroom ontstaat meestal als gevolg van een pijncrisis.
De symptomen van het acute thoraxsyndroom zijn vergelijkbaar met die van longontsteking: de getroffenen (vaak kinderen) hebben koorts, hoesten, ademhalingsmoeilijkheden en hevige pijn op de borst, vooral bij het ademen. Als het thoracale syndroom steeds weer optreedt, beschadigt het de longen op de lange termijn.
Als de getroffenen regelmatig ademhalingsoefeningen en/of ademhalingstherapie uitvoeren, is het mogelijk om een acuut borstsyndroom te voorkomen. De zieken leren oefeningen (bijvoorbeeld rekoefeningen) en technieken die het ademen voor hen gemakkelijker maken.
Als er tekenen zijn van acuut borstsyndroom, is het belangrijk om de persoon onmiddellijk naar het ziekenhuis te brengen!
hartinfarct
Bij sikkelcelziekte is het ook mogelijk dat grotere bloedvaten (bijvoorbeeld in de hersenen) door de sikkelcellen vernauwen of verstopt raken. Als bijvoorbeeld een vat in de hersenen verstopt raakt, wordt de omgeving niet meer voldoende voorzien van zuurstof en voedingsstoffen. Er ontstaat een beroerte.
Tekenen van een beroerte zijn onder meer plotselinge hoofdpijn, plotselinge gevoelloosheid of zwakte in een helft van het gezicht, arm of been of zelfs in het hele lichaam, spraakproblemen of een aanval.
Als de patiënt tekenen van een beroerte vertoont, bel dan onmiddellijk een spoedarts!
Wanneer naar de dokter?
Bepaalde symptomen duiden op ernstige, soms levensbedreigende complicaties bij mensen met sikkelcelanemie. Het is daarom raadzaam om direct een arts te raadplegen als:
- Zieke mensen hebben koorts boven de 38,5 graden Celsius
- ze zijn bleek en slap
- ze hebben pijn op de borst
- je bent kortademig
- Gewrichtspijn treedt op
- ze hebben een opgeblazen gevoel en/of buikpijn
- de milt is voelbaar en vergroot
- Mensen krijgen ineens gele ogen
- ze scheiden zeer donkere urine uit
- ze hebben hoofdpijn, duizeligheid en/of verlamming, gevoelloosheid
- de penis is stijf en pijnlijk (priapisme)
Hoe behandel je sikkelcelziekte?
Als de arts sikkelcelziekte heeft vastgesteld, is het van belang dat de betrokkene wordt behandeld door een gespecialiseerd behandelteam dat nauw samenwerkt met huisartsen en kinderartsen. Zo krijgt de zieke de best mogelijke therapie.
Het doel van de behandeling is het verlichten van symptomen en het voorkomen van eventuele klachten en complicaties. Artsen schrijven hiervoor meestal medicijnen voor (bijvoorbeeld hydroxycarbamide, pijnstillers). In ernstige gevallen voeren artsen stamceltransplantaties uit en in zeldzame gevallen gentherapie.
Hydroxycarbamide
Artsen behandelen patiënten met sikkelcelziekte gewoonlijk met de werkzame stof hydroxycarbamide (ook: hydroxyureum). Het is een cytostaticum dat ook wordt gebruikt om kanker te behandelen. Dit medicijn maakt het bloed vloeibaarder en verhoogt de hoeveelheid foetaal hemoglobine (HbF) in het lichaam. Foetaal hemoglobine wordt aangetroffen bij gezonde pasgeborenen en remt de vorming van sikkelcellen.
Als mensen met sikkelcelanemie regelmatig hydroxycarbamide nemen, treden pijncrises minder vaak op. Het medicijn verhoogt ook het rode bloedpigment hemoglobine in het bloed, waardoor patiënten zich over het algemeen beter voelen.
Pijnstiller
Mensen met sikkelcelziekte ervaren vaak hevige pijn, veroorzaakt door bijvoorbeeld pijncrises, hand-voetsyndroom en zweren. Artsen schrijven daarom pijnstillers voor met de werkzame stoffen paracetamol, metamizol, diclofenac of ibuprofen. De artsen beslissen individueel in welke dosering, waar (in het ziekenhuis of thuis) en in welke vorm (bijvoorbeeld sap, tablet, infuus) de patiënten de medicatie krijgen.
Mensen met sikkelcelanemie mogen geen acetylsalicylzuur (ASA) krijgen. Dit actieve ingrediënt verhoogt het risico op ernstige hersen- en leverschade.
Bloedtransfusie
Artsen schrijven bloedtransfusies voor aan patiënten met sikkelcelanemie in het geval van of om beroertes te voorkomen, evenals bij miltsequestratie en acuut thoracaal syndroom. Deze vinden regelmatig (meestal maandelijks) en vaak levenslang plaats.
Om dit te doen, geven de artsen infusies met erytrocytenconcentraten. Deze bevatten rode bloedcellen van een gezonde bloeddonor. Dit geeft de getroffen persoon de dringend benodigde rode bloedcellen. Tegelijkertijd vervangt de arts sikkelcellen door normale rode bloedcellen om het risico op verdere complicaties (bijvoorbeeld acuut borstsyndroom of een beroerte) te verminderen.
Vaccinaties
Om levensbedreigende infecties te voorkomen, vaccineren artsen jonge kinderen met sikkelcelanemie al in de tweede levensmaand tegen pneumokokken (bacteriën die longontsteking veroorzaken). Andere speciale vaccinaties die artsen aanbevelen voor sikkelcelziekte zijn vaccinaties tegen meningokokken (meningitis), hemophilus (kroep, longontsteking, meningitis, gewrichtsontsteking), griepvirussen en hepatitisvirussen.
antibiotica
Artsen raden aan om kinderen met sikkelcelziekte vanaf de leeftijd van drie maanden tot vijf maanden elke dag penicilline (antibioticum) te geven. Dit helpt ernstige bacteriële infecties te voorkomen, waar mensen met sikkelcelziekte bijzonder vatbaar voor zijn.
Als patiënten met sikkelcelanemie galblaasinfecties, een maagzweer of een infectieziekte krijgen, geven artsen ook antibiotica.
Stamceltransplantatie
Met een stamceltransplantatie (beenmergtransplantatie) kunnen artsen mensen met sikkelcelziekte genezen.
Stamceltransplantatie brengt echter risico's met zich mee en is niet geschikt voor iedereen met sikkelcelziekte. Het wordt daarom alleen uitgevoerd bij patiënten met een zeer ernstig beloop van de ziekte.
Hoe werkt de stamceltransplantatie?
Artsen nemen stamcellen uit het beenmerg van een gezonde donor (donormerg) om gezond bloed te produceren (bloedstamcellen). Vóór de transplantatie gebruiken de artsen chemotherapie of bestralingstherapie om het beenmerg van de patiënt (ontvanger), dat de sikkelcellen vormt, te vernietigen.
De artsen gebruiken dan een bloedtransfusie om de zieke persoon de gezonde stamcellen te geven. Zo vervangen ze het zieke beenmerg door gezonde stamcellen en maakt de getroffen persoon nu zelf nieuwe, gezonde bloedcellen aan.
Om ervoor te zorgen dat het lichaam van de ontvanger de nieuwe stamcellen van de donor accepteert, moet het meeste bloed overeenkomen. Artsen gebruiken daarom liever broers en zussen als donor, omdat ze vaak dezelfde kenmerken (HLA-genen) hebben.
Gentherapie
Artsen doen al enkele jaren onderzoek om vast te stellen of sikkelcelziekte blijvend te genezen is met gentherapie. Hiervoor nemen ze de stamcellen van de aangedane persoon, vervangen het pathologische hemoglobinegen door een gezond gen en voeden de genetisch gemodificeerde stamcellen vervolgens terug in het lichaam van de persoon. Dit is dan in staat om gezonde bloedcellen te produceren.
Tot nu toe is hier echter alleen ervaring mee opgedaan bij individuele patiënten. Nader onderzoek is nodig om te verduidelijken in hoeverre gentherapie kan worden ingezet als behandelmethode voor sikkelcelziekte.
Meer therapieën
Er zijn enkele veelbelovende medicijnen (bijvoorbeeld preparaten met het aminozuur L-glutamine) die onderzoekers momenteel testen en die mogelijk binnen tien jaar op de markt komen. Bovendien ontwikkelen artsen voortdurend stamceltransplantaties. De zogenaamde haploidente stamceltransplantatie, waarbij meestal één ouder als donor dient voor een ziek kind, belooft succes. Haploident betekent dat donor en ontvanger maar half hetzelfde zijn wat betreft HLA-kenmerken.
De behandeling van mensen met sikkelcelziekte is voortdurend in ontwikkeling. Het is daarom belangrijk dat zieke mensen en hun naasten zich regelmatig informeren over actuele therapieën.
Hoe stelt de arts een diagnose?
Het eerste aanspreekpunt bij een vermoeden van sikkelcelziekte is meestal uw kinderarts. Indien nodig en voor nader onderzoek verwijst hij u door naar een specialist interne geneeskunde, gespecialiseerd in bloedziekten (hematoloog).
Aangezien sikkelcelanemie een erfelijke ziekte is, geeft de familiegeschiedenis de eerste aanwijzingen. De arts vraagt bijvoorbeeld of er in de familie dragers van de ziekte zijn.
Artsen raden ook aan om bij alle pasgeborenen uit risicogebieden (bijv. West- en Centraal-Afrika) een bloedonderzoek te laten uitvoeren. Sommige landen doen wat bekend staat als screening van pasgeborenen voor sikkelcelanemie. Een pasgeboren screening is een algemeen onderzoek van alle pasgeborenen om bepaalde aangeboren ziekten in een vroeg stadium op te sporen.
Artsen raden ook aan om oudere kinderen en adolescenten uit risicogebieden te controleren die bloedarmoede en vaak ernstige pijn of infecties voor sikkelcelziekte zijn. Als er aanwijzingen zijn voor de ziekte, zal de arts een bloedonderzoek doen.
Bloed Test
Om sikkelcelanemie te diagnosticeren, zal de arts een bloedtest doen. In het bloeduitstrijkje kan hij de typische sikkelvormige rode bloedcellen meestal direct onder een microscoop zien. Hiervoor neemt hij een druppel bloed van de betrokkene en spreidt deze uit op een drager (bijvoorbeeld op glas). Als er geen sikkelcellen zijn, betekent dit niet dat er geen sikkelcelziekte is.
Een preciezere diagnose van sikkelcelziekte wordt in het laboratorium uitgevoerd met behulp van hemoglobine-elektroforese (Hb-elektroforese). Het is een van de belangrijkste onderzoeksmethoden om aandoeningen van het rode bloedpigment op te sporen. Bij hemoglobine-elektroforese wordt de hemoglobine uit het bloed van de patiënt in een vloeistof opgelost en op een drager aangebracht. Er wordt dan een spanning aangelegd. Afhankelijk van hoe de hemoglobine is samengesteld, legt het in een bepaalde tijd verschillende afstanden af. Dit kan worden gebruikt om te beoordelen of de hemoglobine normaal of defect is.
Als er bijvoorbeeld abnormale hemoglobine (HbS) wordt gevonden, dat minder dan 50 procent van de totale hemoglobine in het bloed uitmaakt, is de patiënt de drager van de ziekte. Als de persoon meer dan 50 procent HbS in zijn totale hemoglobine heeft, is de kans groot dat hij sikkelcelanemie heeft.
Moleculair genetische studies
Pathologische veranderingen (mutaties) in het hemoglobinegen van de getroffen persoon kunnen ook worden vastgesteld met moleculair biologisch onderzoek. Een moleculair bioloog analyseert bijvoorbeeld specifieke gensegmenten die verantwoordelijk zijn voor sikkelcelziekte uit een bloed- of speekselmonster. Hiervoor wordt meestal de polymerasekettingreactie (PCR) gebruikt. Hierbij worden de gensegmenten gekopieerd en onderzocht met behulp van een enzym (polymerase).
Verder onderzoek
Om symptomen van sikkelcelziekte te voorkomen of eventuele complicaties in een vroeg stadium te voorkomen, zijn ook de volgende onderzoeken mogelijk:
- Onderzoek van het kind in de baarmoeder voor de geboorte (prenatale diagnose)
- Regelmatige controles in gespecialiseerde centra
- Bloed- en urineonderzoek
- Onderzoek van het hersenvocht (CSF)
- Bepaling van de bloedgroep als een bloedtransfusie nodig is
- Echografie van de buik en het hart
- Echografie van de bloedvaten in de hersenen (bijvoorbeeld bij vermoeden van een beroerte)
- Röntgenfoto's van de longen, botten of gewrichten
- Computertomografie (CT) of magnetische resonantie beeldvorming (MRI)
- Onderzoek van de ogen (bijv. bij vermoeden van visuele stoornissen)