fenylketonurie
Marian Grosser studeerde humane geneeskunde in München. Daarnaast durfde de arts, die in veel dingen geïnteresseerd was, spannende omwegen te maken: filosofie en kunstgeschiedenis studeren, voor de radio werken en tenslotte ook voor een Netdoctor.
Meer over de experts Alle inhoud van wordt gecontroleerd door medische journalisten.Fenylketonurie (PKU) is een aangeboren, erfelijke ziekte van het eiwitmetabolisme. Het voorkomt de afbraak van het aminozuur fenylalanine. Dit hoopt zich op in het lichaam en verstoort de ontwikkeling van de hersenen van het kind. Indien onbehandeld, leidt fenylketonurie tot ernstige verstandelijke handicaps. Met tijdige therapie kunnen patiënten echter een normaal leven leiden. Lees hier alles over fenylketonurie!
ICD-codes voor deze ziekte: ICD-codes zijn internationaal erkende codes voor medische diagnoses. Ze staan bijvoorbeeld in doktersbrieven of op attesten van arbeidsongeschiktheid. E70
Fenylketonurie: beschrijving
Fenylketonurie (PKU) is een erfelijke stofwisselingsziekte die vanaf de geboorte bestaat en de afbraak van het essentiële aminozuur fenylalanine verstoort. Aminozuren zijn de basisbouwstenen van eiwitten en dus vitale metabolische componenten. Sommige kunnen alleen via voedsel in het lichaam komen, het organisme kan ze niet zelf aanmaken. Dergelijke aminozuren worden essentieel genoemd.
Wat gebeurt er met fenylketonurie?
Normaal gesproken zijn aminozuren onderhevig aan een balans tussen opname/opbouw en afbraak, zodat er altijd zoveel beschikbaar is als het lichaam nodig heeft. Bij verschillende aminozuren kan een tekort of overmaat aanzienlijke schade aanrichten en verschillende symptomen veroorzaken.
Bij de zogenaamde klassieke PKU is de werking van fenylalanine hydroxylase (PAH) beperkt of zelfs helemaal afwezig. Door het PAK-tekort hoopt fenylalanine zich steeds meer op in het lichaam. Een te hoge concentratie fenylalanine verstoort de hersenontwikkeling aanzienlijk en leidt bij jonge patiënten in een vroeg stadium tot verstandelijke beperkingen.
Omdat de normale afbraak van fenylalanine bij de ziekte niet mogelijk is, ontstaan er andere afbraakproducten, de zogenaamde fenylketonen. Ze worden uitgescheiden in de urine en zijn verantwoordelijk voor de naam van de ziekte.
Atypische fenylketonurie
Zelfs bij atypische vormen van fenylketonurie wordt de afbraak van fenylalanine verstoord. De oorzaak is echter niet een defect in de PAK. In plaats daarvan is de functie van een co-enzym, tetrahydrobiopterine (BH4), beperkt. Het is indirect betrokken bij de afbraak van fenylalanine omdat de PAK BH4 nodig heeft om fenylalanine om te zetten in tyrosine.
Omdat BH4 ook belangrijk is voor de aanmaak van de boodschapperstoffen dopamine en serotonine, is atypische fenylketonurie met BH4-tekort meestal gecompliceerder dan de klassieke vorm.
Op wie is fenylketonurie van invloed?
PKU is een van de meest voorkomende aangeboren stofwisselingsziekten. Naar schatting zal ongeveer één op de 7.000 pasgeborenen wereldwijd het ontwikkelen, zonder verschil tussen meisjes en jongens. Omdat het een erfelijke ziekte is, worden vaak meerdere leden van een familie getroffen.
Fenyketonurie: symptomen
Baby's met fenylketonurie vertonen in eerste instantie geen symptomen van de ziekte. De eerste problemen treden pas op in de vierde tot zesde levensmaand, als de ziekte nog niet is herkend en behandeld. Bovenal veroorzaakt de verstoorde hersenrijping in de loop van de tijd enorme complicaties. Symptomen van onbehandelde PKU zijn onder meer:
- een sterke mentale achterstand. De hersenbeschadiging vordert door de puberteit en stagneert dan. De getroffen kinderen zijn dan meestal ernstig verstandelijk gehandicapt.
- Toevallen (aanvallen) (epilepsie). Door de beschadiging zijn de zenuwcellen van de hersenen bijzonder gevoelig en overprikkelbaar. Frequente epileptische aanvallen zijn het gevolg.
- motorische handicaps. Niet alleen de hersencellen, maar ook de spieren van de patiënt kunnen overprikkeld raken. Daarom wordt het vaak gespannen (spasticiteit), wat leidt tot verschillende bewegingsstoornissen.
- Gedragsstoornissen. Sommige kinderen met fenylketonurie zijn hyperactief en ongewoon agressief, en woede-uitbarstingen komen ook vaker voor.
- een klein hoofd (microcefalie). Doordat de hersenen van de patiënt zich niet goed ontwikkelen, blijft ook de hoofdgroei achter. De kleine hoofdomtrek in vergelijking met hun leeftijdsgenoten valt vooral op bij oudere kinderen.
- een merkbare geur. PKU produceert bepaalde afbraakproducten van fenylalanine die vergelijkbaar zijn met uitwerpselen van muizen. Deze stoffen worden voornamelijk via de urine uitgescheiden, maar ook gedeeltelijk via de huid.
- eczeem-achtige huidveranderingen
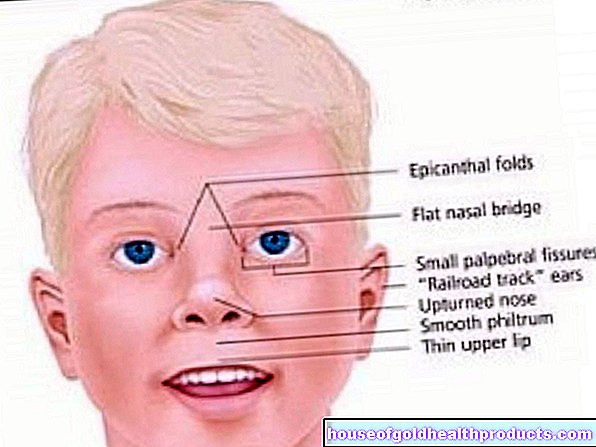
Omdat bij fenylketonurie ook de aanmaak van het pigment melanine wordt verstoord, hebben veel patiënten een zeer lichte, zongevoelige huid en witblond haar. De iris van de ogen is ook lichtblauw tot transparant en laat de roodachtige fundus doorschijnen.
De PKU-symptomen variëren in ernst van persoon tot persoon. De belangrijkste reden hiervoor is dat de activiteit van fenylalaninehydroxylase (PAH) bij elke patiënt anders wordt beperkt. Sommige hebben nog een bepaalde restactiviteit, waardoor er minder fenylalanine in het organisme ophoopt. Anderen vertonen helemaal geen enzymactiviteit - de ziekte vordert dienovereenkomstig sneller en ernstiger.
Fenylketonurie: oorzaken en risicofactoren
Fenylketonurie is een erfelijke ziekte. Inmiddels zijn er tal van genetische mutaties bekend die leiden tot een defect in PAK. Het type mutatie bepaalt in hoeverre de afbraak van fenylalanine wordt beperkt.
PKU wordt recessief overgeërfd, wat betekent dat een persoon drager kan zijn van een veranderd gen zonder de ziekte te ontwikkelen. Evenzo kunnen mensen met fenylketonurie gezonde kinderen verwekken.
Alleen als beide ouders mutaties in hun genetische samenstelling hebben, is er een zekere kans dat hun nakomelingen fenylketonurie zullen ontwikkelen. Als de ouders niet alleen drager zijn van de genen, maar zelf ook PKU hebben, zullen alle kinderen samen dit ook krijgen.
Fenylketonurie: onderzoeken en diagnose
Omdat de ernstige gevolgen van fenylketonurie kunnen worden voorkomen door tijdig met de behandeling te beginnen, is het vooral belangrijk om de ziekte zo vroeg mogelijk te ontdekken. In Duitsland worden kinderen op de derde dag na de geboorte onderzocht op verschillende aangeboren aandoeningen, waaronder PKU, als onderdeel van een algemeen onderzoek (newborn screening).
Tandem massaspectrometrie
Veel aangeboren stofwisselingsstoornissen worden nu gediagnosticeerd met behulp van zogenaamde tandem-massaspectrometrie. Hiermee kan het bloed van de pasgeborene snel en gemakkelijk worden onderzocht. Naast fenylketonurie kunnen artsen binnen enkele minuten meer dan 20 andere ziekten opsporen.
Guthrie-test
De Guthrie-test, genoemd naar de uitvinder, maakt het ook mogelijk om PKU te diagnosticeren. Om dit te doen, wordt een kleine hoeveelheid bloed uit de hiel van het kind genomen en op een stuk filtreerpapier aangebracht. In het laboratorium kun je dan bepalen of de concentratie fenylalanine verhoogd is.
De Guthrie-test werd in de jaren zestig geïntroduceerd en is lange tijd de standaardmethode geweest voor het diagnosticeren van fenylketonurie. Het heeft echter nadelen in vergelijking met tandemmassaspectrometrie. De Guthrie-test levert pas na vijf dagen een resultaat op, dat ook nog eens foutgevoelig is. Factoren zoals het dieet van het kind of mogelijke antibioticatherapie vervalsen bijvoorbeeld de resultaten. In sommige landen wordt de Guthrie-test nog steeds gebruikt, in Duitsland wordt deze meestal niet meer gebruikt.
Andere testen
Als uit de screening van pasgeborenen een vermoeden van fenylketonurie blijkt, volgt nader onderzoek ter bevestiging. Dit kan ook worden gebruikt om de exacte concentratie van fenylalanine in het bloed te bepalen.
Tot slot is het belangrijk om te onderscheiden of het een typische (klassieke) of atypische PKU is. Hiervoor zijn ook speciale tests beschikbaar, zoals de tetrahydrobiopterine-stresstest. Het onderscheid is belangrijk omdat atypische fenylketonurie anders wordt behandeld dan de klassieke vorm.
Vruchtwateronderzoek
Fenylketonurie kan al tijdens de zwangerschap worden vastgesteld (prenatale diagnostiek). Om dit te doen, wordt een kleine hoeveelheid vruchtwater uit de vruchtzak van de moeder gehaald en worden de cellen van het ongeboren kind daarin onderzocht. Eventuele genetische defecten die PKU veroorzaken, kunnen op deze manier worden geïdentificeerd.
Bij pasgeboren screening wordt fenylketonurie echter meestal vroeg genoeg ontdekt om te worden behandeld. Omdat een vruchtwateronderzoek altijd gepaard gaat met een bepaald risico, is het gebruik ervan voor het diagnosticeren van PKU meestal niet zinvol.
Fenylketonurie: behandeling
Er is maar één manier om het teveel aan fenylalanine in PKU tegen te gaan: De getroffen kinderen moeten een speciaal dieet volgen om zo min mogelijk fenylalanine via hun voedsel binnen te krijgen. Ze hebben ook bepaalde voedingssupplementen nodig ter vervanging van stoffen die bij gezonde kinderen uit fenylalanine zouden worden gevormd.
De therapie moet beginnen voordat de eerste symptomen van een ontwikkelingsstoornis optreden, dus binnen de eerste twee levensmaanden. Hersenschade die al is opgetreden, kan niet worden teruggedraaid.
Voedingstherapie met het PKU-dieet
Alle natuurlijke eiwitten bestaan voor ongeveer vijf procent uit het aminozuur fenylalanine. De meeste voedingsmiddelen bevatten veel meer dan het lichaam nodig heeft. Dit is geen probleem voor een gezond persoon omdat hij de overtollige hoeveelheden fenylalanine afbreekt en uitscheidt.
Patiënten met fenylketonurie daarentegen moeten het grotendeels zonder natuurlijke eiwitten stellen. Industrieel vervaardigde speciale producten vervangen de ontbrekende voedselcomponenten. Enerzijds wil men een overmaat aan fenylalanine voorkomen, anderzijds moet de patiënt worden voorzien van stoffen die anders ontbreken, zoals tyrosine, dat wordt gevormd uit fenylalanine.
Het doel van het PKU-dieet is echter niet om de opname van fenylalanine volledig te stoppen. Omdat het organisme een bepaalde hoeveelheid van het aminozuur nodig heeft voor belangrijke stofwisselingsprocessen. Het bereiken van de juiste concentratie is een grote uitdaging voor artsen en patiënten en vereist veel discipline.
Het dieet moet daarom een leven lang worden gevolgd, maar vooral strikt tot de leeftijd van zes jaar. Omdat de hersenen zich tot op deze leeftijd zeer sterk ontwikkelen en daardoor bijzonder gevoelig zijn voor beschadiging. Op volwassen leeftijd veroorzaken hoge concentraties fenylalanine geen hersenbeschadiging zoals bij kinderen. Ze kunnen echter de trigger zijn voor andere neurologische klachten, zoals een slechte concentratie of vertraagde reacties.
Dieetbehandeling voor fenylketonurie moet beginnen bij een gespecialiseerd centrum voor metabole ziekten. Omdat het niet in elke kliniek mogelijk is om ouders te instrueren over het dieet. Dit kan het beste worden gedaan met behulp van dieetadvisering, die laat zien hoe u met het dieet omgaat en regelmatig de fenylalaninespiegels in het bloed controleert.
Behandeling van atypische fenylketonurie
Bij atypische vormen van PKU worden het ontbrekende co-enzym BH4 en bepaalde boodschapperstoffen zoals dopamine en serotonine kunstmatig vervangen. In sommige gevallen moet de patiënt ook een fenylalaninearm dieet volgen.
Fenylketonurie tijdens de zwangerschap
Als u zwanger bent en zelf fenylketonurie heeft, moet u rekening houden met het volgende:
- Houd u bijzonder strikt aan uw dieet!
- Laat regelmatig uw fenylalanine bloedwaarden controleren. Omdat de concentraties van fenylalanine bij het ongeboren kind ongeveer twee keer zo hoog zijn als bij de zwangere vrouw. Naast de hersenen kunnen ze ook het hart en de ogen van het ongeboren kind beschadigen. Misvormingen van het skelet van het kind kunnen ook het gevolg zijn van hoge fenylalaninespiegels tijdens de zwangerschap.
- Om hersenbeschadiging bij het kind in de vroege zwangerschap uit te sluiten, moeten vrouwen met fenylketonurie de zwangerschap zorgvuldig plannen en vanaf het begin voorzorgsmaatregelen nemen.
- Bij zwangere vrouwen met fenylketonurie kan een niet strikt gevolgd dieet leiden tot een miskraam (spontane abortus).
- In ieder geval dienen zwangere vrouwen met fenylketonurie advies en begeleiding in te winnen bij een arts.
Fenylketonurie: ziekteverloop en prognose
Als fenylketonurie vroeg wordt gediagnosticeerd, zo mogelijk bij de pasgeborene, en als het speciale PKU-dieet wordt gevolgd, is de prognose meestal goed. De kinderen ontwikkelen zich geestelijk en hebben een gemiddelde levensverwachting.
Indien onbehandeld, leidt de hersenbeschadiging echter tot ernstige psychische ontwikkelingsstoornissen die later niet kunnen worden gecorrigeerd. De getroffenen hebben een normale levensverwachting, maar hun intelligentiequotiënt ligt bijna altijd ver onder de norm.
De zeldzame atypische vorm van fenylketonurie, waarbij BH4-deficiëntie aanwezig is, is een speciaal geval. Deze variant van fenylketonurie kan ondanks een dieet leiden tot progressieve neurologische schade met ernstige krampen.
Tags: zwangerschap digitale gezondheid tcm