Chorea Huntington
Ingrid Müller is chemicus en medisch journalist. Ze was twaalf jaar hoofdredacteur van . Sinds maart 2014 werkt ze als freelance journalist en auteur voor Focus Gesundheit, het gezondheidsportaal ellviva.de, de uitgeverij living crossmedia en het gezondheidskanaal van rtv.de.
Meer over de experts Alle inhoud van wordt gecontroleerd door medische journalisten.De ziekte van Huntington (de ziekte van Huntington, verouderd: St. Vitus's Dance) is een erfelijke hersenziekte. Bij mensen met de ziekte van Huntington worden hersengebieden die belangrijk zijn voor spiercontrole en psychologisch functioneren geleidelijk vernietigd. De zenuwcellen sterven langzaam af. De oorzaak is een defect gen Lees meer over de ziekte van Huntington.
ICD-codes voor deze ziekte: ICD-codes zijn internationaal erkende codes voor medische diagnoses. Ze staan bijvoorbeeld in doktersbrieven of op attesten van arbeidsongeschiktheid. G10

Ziekte van Huntington: beschrijving
De ziekte van Huntington is een zeer zeldzame erfelijke hersenziekte die ongeveer zeven tot tien op de 100.000 mensen treft in West-Europa en Noord-Amerika. In Duitsland lijden ongeveer 8.000 mensen aan de ziekte van Huntington.
De ziekte van Huntington kan op elke leeftijd uitbreken. De eerste symptomen van de ziekte van Huntington treden vaak op tussen het 35e en 45e levensjaar, maar ze kunnen ook in de vroege kinderjaren of op hoge leeftijd optreden. Dit hangt ook af van waar precies de ziekteveroorzakende verandering (mutatie) in de genetische samenstelling ligt. Typische symptomen zijn bewegingsstoornissen en karakterveranderingen, waaronder dementie.
De mogelijkheden om het verloop van de chorea van Huntington te beïnvloeden zijn beperkt. Er is een groot aantal stoffen getest die de zenuwcellen beschermen en de voortschrijdende neuronale vernietiging stoppen. Tot nu toe heeft echter geen van deze stoffen een significant effect gehad op het ziekteverloop. Er zijn echter enkele medicijnen beschikbaar die de symptomen van de ZvH kunnen helpen verminderen.
Er zijn steungroepen die mensen met de ziekte van Huntington en hun dierbaren helpen.
Waar de naam "ziekte van Huntington" vandaan komt?
De chorea van Huntington gaat terug op de Amerikaanse arts George Huntington, die de ziekte voor het eerst beschreef in 1872. Hij realiseerde zich ook dat de ziekte van Huntington erfelijk is. De naam komt van het Grieks - "choreia" = "dans". De ziekte van Huntington heeft verschillende namen: de ziekte van Huntington, de ziekte van Huntington of, in het verleden, St. De reden is dat patiënten geleidelijk de controle over hun bewegingen verliezen - wat vaag kan doen denken aan een dans.
Ziekte van Huntington: symptomen
Ziekte van Huntington: vroege stadia
De ziekte van Huntington begint vaak met niet-specifieke symptomen, zoals psychische problemen, die toenemen. Veel patiënten zijn prikkelbaarder, agressiever, depressiever of ongeremd; anderen voelen een verlies van spontaniteit of een toenemende angst.
De bewegingsstoornissen bij de ziekte van Huntington bestaan meestal uit plotselinge, onwillekeurige bewegingen van hoofd, handen, armen, benen of romp. In extreme gevallen kan dit leiden tot een steigerende gang die typisch is voor de ziekte. Daarom werd de ZvH in het verleden ook wel "St. Vitusdans" genoemd.
Mensen met de ziekte van Huntington kunnen deze overdreven en ongewenste bewegingen aanvankelijk vaak in hun bewegingen opnemen. Voor de toeschouwer wekt dit de indruk van overdreven gebaren. In veel gevallen merken de getroffenen hun bewegingsstoornissen in eerste instantie niet als zodanig op.
Ziekte van Huntington: latere stadia
Naarmate de ziekte van Huntington vordert, worden ook de tong- en keelspieren in toenemende mate aangetast. De taal lijkt schokkerig, geluiden worden explosief geuit. Slikstoornissen zijn ook mogelijk - dan bestaat het risico dat de getroffenen voedselcomponenten doorslikken en longontsteking krijgen.
Naarmate de ziekte van Huntington vordert, verliezen patiënten vaak onverbiddelijk hun mentale vermogens. Na ongeveer 15 jaar kan bij bijna alle getroffenen dementie worden vastgesteld.
In de laatste stadia van de ziekte van Huntington zijn patiënten meestal bedlegerig en volledig afhankelijk van de hulp van anderen.
Ziekte van Huntington - achtige ziekten
Symptomen die vergelijkbaar zijn met die van de ziekte van Huntington kunnen ook worden veroorzaakt door andere, niet-erfelijke oorzaken. Voorbeelden zijn de gevolgen van infectieziekten of hormonale veranderingen tijdens de zwangerschap (chorea gravidarum). Medicijnen en hormonale anticonceptiva (zoals de pil) zijn minder vaak voorkomende triggers. Dit geldt ook voor beroertes die bepaalde hersengebieden aantasten. Door stoornissen in de bloedsomloop in de hersenen kan zelfs op oudere leeftijd een vorm van chorea ontstaan. Er wordt aangenomen dat ziekten zoals hyperthyreoïdie ook tot chorea kunnen leiden. Het beloop is hier echter niet progressief en de bewegingsstoornissen verdwijnen meestal weer. Ernstige psychische symptomen zijn in dergelijke gevallen atypisch.
De ziekte van Huntington: oorzaken en risicofactoren
De ziekte van Huntington wordt veroorzaakt door de genetische dood van zenuwcellen in bepaalde delen van de hersenen. De ziekte kan zowel mannen als vrouwen treffen, omdat het verantwoordelijke gen niet geslachtsgebonden wordt geërfd - dus niet op de geslachtschromosomen X en Y, maar op de autosomen. Dit zijn die chromosomen die het geslacht niet bepalen en die in twee exemplaren beschikbaar zijn (een van de moeder, een van de vader).
Elk kind van een ouder die het defecte gen draagt, heeft 50 procent kans om het defecte gen te krijgen en de ZvH te ontwikkelen. Experts spreken van een autosomaal dominante overerving. Dominant betekent dat de ziekte uitbreekt, zelfs als het overeenkomstige gen op slechts één van de twee gepaarde chromosomen wordt veranderd.
Overerving - fout op chromosoom vier
Het moleculaire alfabet van het genetisch materiaal (deoxyribonucleïnezuur, kort DNA of DNA) bestaat uit vier nucleïnezuren: adenine, thymine, guanine en cytosine. Nieuwe combinaties van deze vier letters vormen de gehele genetische informatie, die is opgeslagen in de vorm van draadachtige structuren, de zogenaamde chromosomen.
Bij de ziekte van Huntington is één gen op chromosoom vier veranderd (ook wel het Huntington-gen genoemd). Het werd in 1993 geïdentificeerd. Het eiwit dat door dit gen wordt gecodeerd, werkt niet goed, wat uiteindelijk leidt tot symptomen van de ZvH.
Bij gezonde mensen herhalen de drie nucleïnezuren cytosine-adenine-guanine zich 10 tot 30 keer in dit gebied op chromosoom vier. Met meer dan 36 cytosine-adenine-guanine-herhalingen (CAG-herhalingen of CAG-tripletten) breekt de ZvH uit. 30 tot 35 CAG-herhalingen worden beschouwd als het grijze gebied.
Bij ongeveer één tot drie procent van degenen met de ziekte van Huntington wordt geen bloedverwant met de ziekte van Huntington gevonden. Ofwel is het een geheel nieuwe genverandering, een zogenaamde nieuwe mutatie. Of een ouder van de ZvH-patiënt had 30 tot 35 herhalingen en kreeg de ziekte niet. In de volgende generatie neemt het aantal herhalingen vaak toe. Meer dan 36 kinderen zullen de ziekte van Huntington ontwikkelen.
Hoe meer CAG-herhalingen op chromosoom vier worden geteld, hoe eerder de ZvH uitbreekt en hoe sneller de ziekte vordert.
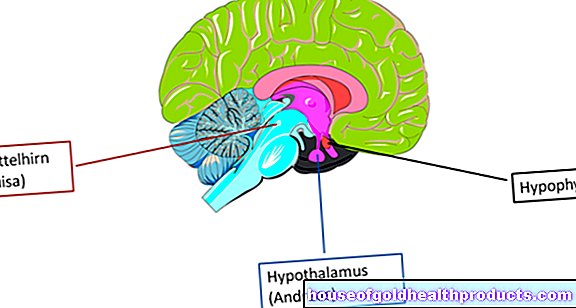
De dood van zenuwcellen bij de ziekte van Huntington beïnvloedt de hersenschors en vooral de zogenaamde basale ganglia. Dit zijn grote groepen zenuwcellen in de twee hersenhelften, die onder andere een rol spelen in de gecoördineerde bewegingsvolgorde.
Ziekte van Huntington: onderzoeken en diagnose
Als de ziekte van Huntington wordt vermoed, is het het beste om een gespecialiseerd centrum voor de ziekte van Huntington te raadplegen met de juiste ervaren neurologen. Omdat huisartsen en soms "normale" neurologen nog nooit een patiënt met deze ziekte hebben gezien.
Aan het begin van de diagnose van de ziekte van Huntington is er een gedetailleerd overzicht, inclusief familiegeschiedenis (anamnese). Voor de arts is het interessant of naaste familieleden (ouders, grootouders) de ZvH hebben. Ook belangrijk voor de diagnose zijn de symptomen van de ZvH (in een vroeg stadium kunnen ze ook wijzen op andere ziekten), het ziekteverloop en afwijkingen bij het neurologisch onderzoek.
De diagnose wordt bevestigd door een bloedonderzoek. Het excessieve aantal herhalingen van dezelfde basenparen (CAG) in een bepaald gen op chromosoom vier kan worden opgespoord door middel van moleculair genetisch onderzoek.
Als er geen genetische verandering kan worden gevonden, wordt opnieuw bloed afgenomen om ziekten uit te sluiten die in eerste instantie symptomen kunnen vertonen die lijken op de ziekte van Huntington. Er worden onder andere schildklierwaarden en de concentratie van bepaalde auto-antistoffen (antistoffen die gericht zijn tegen lichaamseigen stoffen) in het bloed bepaald.
Bepaal zenuwbeschadiging
Om de omvang van zenuwbeschadiging te bepalen, worden mensen met de ziekte van Huntington ook neurologisch, neuropsychologisch en psychiatrisch onderzocht. Deze onderzoeken worden uitgevoerd door ervaren artsen of speciaal opgeleide neuropsychologen.
Beeldvormende onderzoeken zoals computertomografie (CT) of magnetische resonantiebeeldvorming (MRI) van de hersenen kunnen de afbraak van individuele hersengebieden (bijvoorbeeld de zogenaamde staartkern en de hersenschors) laten zien. Elektrofysiologische diagnostiek kan ook in individuele gevallen belangrijke informatie opleveren over de ZvH.
Genetische tests voor de ziekte van Huntington
Zelfs gezonde mensen met een verhoogd risico op de ziekte van Huntington kunnen een genetische test krijgen. Artsen spreken van voorspellende diagnostiek of voorspellende diagnostiek. Deze test kan worden gebruikt om duidelijk te bepalen of iemand de ZvH zal krijgen of niet.
Deze kennis van de drager te zijn van deze genetische verandering kan echter een enorme impact hebben op de psyche van de getroffenen. Als gevolg hiervan zijn er richtlijnen voor het verkrijgen van een genetische test voor de ziekte van Huntington. Betrokkenen dienen onder meer vooraf geïnformeerd te worden over de risico's. Er mogen geen genetische tests worden uitgevoerd op minderjarigen. Ook niet bij personen op verzoek van derden, bijvoorbeeld artsen, verzekeringsmaatschappijen, adoptiebureaus of werkgevers.
Ziekte van Huntington: behandeling
Geneesmiddelen tegen de ziekte van Huntington
De ziekte van Huntington kan niet causaal worden behandeld en is niet te genezen. Bepaalde medicijnen verlichten echter de symptomen. De actieve ingrediënten tiapride en tetrabenazine kunnen overmatige en ongecontroleerde bewegingen beperken door de lichaamseigen boodschapperstof dopamine tegen te gaan. Bij depressieve stemmingen kunnen antidepressiva uit de groep van selectieve serotonineheropnameremmers (SSRI's) of sulpiride de symptomen verminderen.
Er is geen officiële therapieaanbeveling tegen de ontwikkeling van dementie bij de ziekte van Huntington. In sommige onderzoeken kon de werkzame stof memantine de mentale achteruitgang vertragen.
Naast medicamenteuze therapie voor de ziekte van Huntington, kunnen ondersteunende maatregelen zoals fysiotherapie, ergotherapie en logopedie ook helpen de symptomen te verlichten. Spraak- of slikstoornissen kunnen bijvoorbeeld worden verbeterd door speciale oefeningen. Daarnaast kan ergotherapie worden gebruikt om de activiteiten van het dagelijks leven te trainen. Hierdoor kunnen mensen met de ziekte van Huntington langer zelfstandig blijven. Psychotherapie helpt veel patiënten om beter met de ziekte om te gaan. Zelfhulpgroepen zijn ook een grote steun voor velen.
Omdat veel mensen met de ZvH aanzienlijk afvallen in de loop van hun ziekte, is een calorierijk dieet zinvol. In sommige gevallen verbetert licht overgewicht de symptomen van de ziekte van Huntington.
Onderzoek naar de ziekte van Huntington
Therapieën die het onderliggende afbraakproces bij de ZvH kunnen stoppen, zijn nog niet bekend. Er worden verschillende nieuwe benaderingen nagestreefd in op medicijnen gebaseerde therapie voor de ziekte van Huntington, maar deze bevinden zich nog in de experimentele fase. Studies hebben aangetoond dat hoge doses CoEnzyme Q10 licht positieve effecten hebben, maar over het algemeen hebben ze geen invloed op het verloop van de ziekte. CoEnzyme Q10 is een eiwit dat van nature in het lichaam voorkomt en de cellen beschermt tegen beschadiging.
Een ander actief ingrediënt dat wordt gebruikt voor de behandeling van de ziekte van Huntington is ethyl icosapent, een geneesmiddel dat speciaal is ontwikkeld voor de ziekte van Huntington en dat is gemaakt van de visoliecomponent eicosapentaeenzuur. Ethyl-Icosapent is bedoeld om schade aan bepaalde celcomponenten die een rol spelen bij celdood te voorkomen en om de efficiëntie van de zieke zenuwcellen te verhogen. Ook bij de bewegingsstoornissen kon dit middel geen verbetering brengen.
Ziekte van Huntington: ziekteverloop en prognose
De ziekte van Huntington is niet te genezen. De getroffenen overlijden gemiddeld 19 jaar na het begin van de ziekte van Huntington. Dit is slechts een gemiddelde waarde, waardoor ook individuele mensen veel later of eerder kunnen overlijden aan de gevolgen van de ZvH.
De symptomen die de getroffenen ontwikkelen en hoe goed ze reageren op de verschillende medicijnen varieert sterk. Alleen specialisten die dagelijks met de ziekte van Huntington te maken hebben, zijn voldoende bekend met de vele eigenaardigheden van de ziekte. Het is daarom raadzaam voor ZvH-patiënten om zich regelmatig te laten controleren in een van de ZvH-centra in Duitsland om de individuele therapie aan te passen aan het huidige ziekteverloop.
Voor familieleden en levenspartners van mensen met de ziekte van Huntington is de ziekte emotioneel, soms ook financieel, erg belastend. Deutsche Huntington Hilfe e.V. is beschikbaar voor families van getroffenen door waardevol advies en belangrijke adressen te verstrekken indien nodig.
Tags: eetpatroon gezonde werkplek baby peuter







.jpg)