Spinale musculaire atrofie
en Florian Tiefenböck, dokter Bijgewerkt opMaximilian Reindl studeerde scheikunde en biochemie aan de LMU in München en is sinds december 2020 lid van de-redactie. Hij maakt zich voor u vertrouwd met medische, wetenschappelijke en gezondheidsbeleidsthema's om ze begrijpelijk en begrijpelijk te maken.
Meer berichten van Maximilian ReindlFlorian Tiefenböck studeerde humane geneeskunde aan de LMU München. Hij kwam in maart 2014 als student bij en ondersteunt sindsdien de redactie met medische artikelen. Na het behalen van zijn medische licentie en praktijkwerk in de interne geneeskunde aan het Universitair Ziekenhuis Augsburg, is hij sinds december 2019 een vast lid van het-team en zorgt hij onder meer voor de medische kwaliteit van de-tools.
Meer berichten van Florian Tiefenböck Alle inhoud van wordt gecontroleerd door medische journalisten.
Spinale musculaire atrofie, of kortweg SMA, is een zeldzame ziekte waarbij bepaalde zenuwcellen in het ruggenmerg afsterven. Prikkels en impulsen uit de hersenen bereiken dan niet meer hun bestemming: de spieren. Dit veroorzaakt spierverlies en verlamming. Er zijn verschillende vormen van SMA. De meest ernstige begint in de kindertijd. Nieuwe behandelingen beloven een blijvende verbetering van de gezondheid. Lees hier meer over spinale musculaire atrofieën.
ICD-codes voor deze ziekte: ICD-codes zijn internationaal erkende codes voor medische diagnoses. Ze staan bijvoorbeeld in doktersbrieven of op attesten van arbeidsongeschiktheid. G12

Kort overzicht
- Wat is spinale musculaire atrofie? Een groep spierzwakte ziekten. Ze zijn gebaseerd op de dood van bepaalde zenuwcellen in het ruggenmerg die de spieren aansturen (motorneuronen). Daarom behoren de SMA tot de motorneuronziekten.
- Welke vormen zijn er? Erfelijke spinale musculaire atrofieën zijn meestal SMA met een bepaald genetisch defect op chromosoom 5 (5q-geassocieerde SMA). Artsen onderscheiden vier verschillende vormen: SMA type 1 - type 4. Daarnaast zijn er sporadische vormen waarvan de erfelijkheid niet zeker is.
- Frequentie: Zeldzame ziekte; de erfelijke SMA treft ongeveer één pasgeborene op 7000.
- Symptomen: spiertrekkingen, progressieve spierzwakte, spierafbraak, verlamming. Verlopen verschillen afhankelijk van de SMA-vorm.
- Oorzaken: De erfelijke spinale musculaire atrofie type 1-4 is het gevolg van een genetisch defect op chromosoom 5, meer bepaald op het SMN1-gen. Hierdoor mist het lichaam een speciaal eiwit, het SMN-eiwit. Dit tekort beschadigt de motorneuronen in het ruggenmerg.
- Diagnose: Genetisch onderzoek voor veranderde SMN-genetische samenstelling, lichamelijk onderzoek, elektroneurografie, elektromyografie, bloedonderzoek (bijv. CK)
- Behandeling: Genvervangende therapie of medicijntoediening van splitsingsmodulatoren zijn mogelijk. Begeleidende fysiotherapie, logopedie, pijntherapie en psychotherapie. Indien nodig, operatie aan de wervelkolom. Behandelplan afhankelijk van het type SMA.
- Prognose: Bij erfelijke proximale SMA hebben nieuwe therapiemogelijkheden een causaal effect en kunnen ze een positief effect hebben op het ziekteverloop. Vroeg beginnen met de behandeling is cruciaal. Behandelingen zijn nog niet voor elke patiënt beschikbaar. Indien onbehandeld, overlijden kinderen met SMA type 1 meestal binnen de eerste twee jaar. Levensverwachting bij type 3 en type 4 niet of nauwelijks verminderd.
Wat is spinale musculaire atrofie?
Bij spinale musculaire atrofie (SMA) sterven bepaalde zenuwcellen in het ruggenmerg af. Ze controleren meestal spieren, daarom verwijzen experts naar deze zenuwcellen als motorneuronen. Dienovereenkomstig behoren de SMA tot de zogenaamde motorneuronziekten.
In het geval van spinale musculaire atrofieën worden de lagere (tweede) motorneuronen aangetast, die met hun aanhangsels direct verbonden zijn met de spieren. Als gevolg van de beschadiging bereiken er minder of geen zenuwsignalen meer de spieren. De spieren worden steeds zwakker en kleiner (spierverspilling / spieratrofie).
Artsen maken onderscheid tussen verschillende vormen van spinale musculaire atrofie. Veruit de grootste groep zijn de erfelijke SMA, waarbij de spieren dichtbij de romp (proximaal) zijn aangetast. Ze zijn gebaseerd op een specifiek genetisch defect. Ongeveer één op de 7.000 pasgeborenen zal het ontwikkelen.
Spinale musculaire atrofie is over het algemeen een zeldzame ziekte. Niettemin is het de op één na meest voorkomende autosomaal recessieve erfelijke ziekte en de meest voorkomende doodsoorzaak bij baby's of peuters als gevolg van een genetisch defect.
Welke soorten spinale musculaire atrofie zijn er?
Artsen onderscheiden erfelijke (erfelijke) vormen van SMA van sporadische vormen. Een andere classificatie van spinale musculaire atrofie heeft voornamelijk betrekking op de spiergroepen die het eerst worden aangetast. Er zijn
- Proximale SMA: Met ongeveer 90 procent vormen ze de grootste SMA-groep. De symptomen beginnen in de spieren nabij de romp, d.w.z. proximaal.
- Niet-proximale SMA: hier worden meer afgelegen spiergroepen, zoals de handen en voeten, het eerst aangetast (distale SMA). In het verdere verloop kunnen deze SMA zich ook verspreiden naar spieren nabij het midden van het lichaam.
- Speciale vormen (bijv. Kennedy-type spinobulbaire spieratrofie)
Proximale spinale musculaire atrofieën
Erfelijke proximale spinale musculaire atrofieën zijn meestal ziekten die zijn gebaseerd op een specifiek genetisch defect (5q-geassocieerde SMA, defect op chromosoom 5). Deze zijn op hun beurt weer onderverdeeld in vier verschillende vormen. De indeling is gebaseerd op het tijdstip waarop de eerste symptomen optreden en op het verloop van de ziekte.
Spinale spieratrofie type 1: Dit is de meest voorkomende en ernstigste vorm van SMA. Het wordt ook wel "ziekte van Werdnig-Hoffmann" of "acute infantiele SMA" genoemd. De ziekte begint meestal in de vroege kinderjaren. De spierzwakte treft het hele lichaam - artsen spreken ook van een "floppy infant syndrome" (uit het Engels floppy = slap, zuigeling = zuigeling, kind). De meeste onbehandelde kinderen met SMA type 1 overlijden voordat ze twee jaar oud zijn.
Spinale musculaire atrofie type 2: Deze vorm van SMA staat ook bekend als "intermediaire spinale musculaire atrofie" of "chronische infantiele SMA". De eerste symptomen verschijnen meestal vóór de leeftijd van 18 maanden. De getroffenen hebben een soms aanzienlijk kortere levensverwachting.
Spinale musculaire atrofie type 3: Het is ook bekend als "juveniele spinale musculaire atrofie" of "ziekte van Kugelberg-Welander". Deze SMA begint meestal na de leeftijd van 18 maanden en vóór de vroege volwassenheid. De spierzwakte is milder dan bij type 1 of 2. De getroffenen hebben slechts een iets kortere levensverwachting.
Spinale musculaire atrofie type 4: Het is vergelijkbaar met SMA type 3, maar verschijnt alleen op volwassen leeftijd (meestal> 30 jaar). De spierzwakte is echter minder uitgesproken en vordert langzamer dan bij SMA type 3.
De overgangen tussen de verschillende versies zijn vloeiend. In sommige gevallen maakt dit een duidelijke afbakening moeilijk. Sommige genetische aanleg spelen ook een belangrijke rol bij de ernst van de ziekte in kwestie.
Andere spinale musculaire atrofieën
Naast deze proximale vormen zijn er nog andere vormen van spinale musculaire atrofie. Deze omvatten bijvoorbeeld zeldzamere, ook erfelijke distale spinale musculaire atrofieën. Bij hen beginnen de symptomen meestal in spiergroepen die verder van het lichaam verwijderd zijn.
Overerving is niet gegarandeerd in het geval van sporadische SMA. Bovendien kan er geen familiale accumulatie worden vastgesteld. In de literatuur zijn dit onder meer:
- Hirayama-type (juveniele distale SMA, ziekte rond de leeftijd van 15, tast armspieren aan, komt meestal tot stilstand, zelfs zonder therapie en kan zelfs verbeteren)
- Vulpian-Bernhard-type (ook "flail-arm"-syndroom met aanvang in de schoudergordel, meestal vanaf de leeftijd van 40 jaar)
- Duchenne-Aran-type (aanvankelijk aangetaste handspieren, verspreidend naar de romp van het lichaam, meestal na de leeftijd van 30)
- Peroneaal type ("flail-leg"-syndroom, eerst op de onderbeenspieren)
- Progressieve bulbaire parese (spraak- en slikstoornissen, treft ongeveer 20 procent van de patiënten met amyotrofische laterale sclerose)
Sommige sporadische SMA-vormen ("flail-arm - / - leg"-syndroom, progressieve bulbaire verlamming) worden in gespecialiseerde kringen tot de varianten van amyotrofische laterale sclerose (ALS) gerekend. Dit artikel gaat voornamelijk over erfelijke proximale spinale musculaire atrofieën.
Spinobulbaire spieratrofie
Spinobulbaire of bulbospinale spieratrofie (Kennedy-type, Kennedy-syndroom) is een erfelijke ziekte. Het begint vaak in de jonge tot midden volwassenheid. Deze speciale SMA-vorm wordt X-gebonden recessief overgeërfd en treft daarom alleen mannen (aangezien mannen maar één X-chromosoom hebben, overheerst bij vrouwen het tweede, gezonde X-chromosoom en zou het defect compenseren).
Spierzwakte in de spieren dicht bij het lichaam op de benen en armen of schouders, evenals de tong- en keelspieren komt vaak voor. Hierdoor hebben getroffenen bijvoorbeeld problemen met spreken en slikken. Ze klagen ook over trillingen, spierkrampen en spiertrekkingen. Getroffen mannen hebben ook vaak onvolgroeide testikels en zijn steriel. Bovendien worden de borstklieren groter (gynaecomastie).
Spinobulbaire spieratrofie is meestal traag. De levensverwachting is nauwelijks beperkt.
Hoe herken je spinale musculaire atrofie?
Typisch voor spinale musculaire atrofie zijn progressieve spierzwakte tot verlamming (parese) en spiertrekkingen. Als gevolg van de zenuwbeschadiging krijgen de spieren geen elektrische impulsen meer, waardoor ze na verloop van tijd krimpen (spieratrofie). De exacte tekenen en klachten zijn afhankelijk van de betreffende vorm. In het volgende gedeelte wordt ingegaan op de symptomen van erfelijke proximale SMA.
Symptomen van infantiele spinale musculaire atrofie Type 1
Bij SMA type 1 verschijnen de symptomen in de eerste zes levensmaanden. Gegeneraliseerde spierzwakte treedt op - dat wil zeggen, een die het hele lichaam beïnvloedt. Daarnaast neemt de spanning tussen de spieren af. Artsen spreken van hypotonie.
Bij pasgeborenen manifesteert deze spierzwakte zich aanvankelijk in een typische beenhouding, die doet denken aan een liggende kikker (kikkerbeenhouding). De benen zijn gebogen, de knieën naar buiten gekanteld en de voeten naar binnen gekanteld. Zelfs het hoofd alleen optillen of vasthouden is meestal niet mogelijk.
Op gevorderde leeftijd kunnen kinderen met SMA type 1 niet zelfstandig zitten of lopen. Veel kinderen kunnen ook niet praten, omdat ook de tongspieren kunnen worden aangetast.
Een ander kenmerk van type 1 spinale musculaire atrofie is de vorm van het bovenlichaam: de spieren van de borst en rug ontwikkelen zich niet goed. Hierdoor krijgt het bovenlichaam een klokvorm (klokborst). Door de lage ontwikkeling van de spieren in borst en rug nemen de getroffenen een voorovergebogen houding aan.
Vaak is er ook een toenemende kromming van de wervelkolom (scoliose). De voorovergebogen en gebogen houding veroorzaakt verdere ademhalingsproblemen. Zeer snelle en oppervlakkige ademhaling (tachypneu) is kenmerkend.
Symptomen van intermediaire spinale musculaire atrofie type 2
Type 2 spinale musculaire atrofie veroorzaakt meestal alleen symptomen tussen de leeftijd van zeven en 18 maanden. Getroffen kinderen kunnen zelfstandig zitten, maar leren meestal niet staan of lopen. De spierzwakte vordert langzamer dan bij type 1.
Bij SMA type 2 treden in de loop van de tijd symptomen op die vergelijkbaar zijn met die van de ernstige infantiele vorm, zoals vervorming van de wervelkolom. De gewrichten verstijven door verkorte spieren en pezen (contracturen). Andere symptomen zijn trillingen in de handen en spiertrekkingen in de tong.
Symptomen van juveniele spinale spieratrofie type 3
Type 3 spinale musculaire atrofie treedt meestal op na de leeftijd van 18 maanden en vóór de leeftijd van 18. Getroffen kinderen kunnen zelfstandig zitten, staan en lopen. De spierzwakte, vooral in de bekken- en beenspieren, veroorzaakt echter een waggelende gang.
In de loop van meerdere jaren nemen de prestaties af: in het begin vinden de getroffenen het moeilijk om te sporten of traplopen, maar uiteindelijk is het ook moeilijk om bijvoorbeeld boodschappentassen te dragen. Na vele jaren maakt type 3 spinale musculaire atrofie hardlopen en elke andere inspanning moeilijk of zelfs onmogelijk, zelfs bij oudere mensen.
Over het algemeen zijn de symptomen echter minder uitgesproken dan bij de twee andere vormen van ziekte, type 1 en type 2. Voor veel van de getroffenen wordt de kwaliteit van leven gedurende een lange periode nauwelijks verminderd.
Symptomen van volwassen spinale musculaire atrofie type
Deze zeer zeldzame vorm van progressieve spierafbraak begint op volwassen leeftijd, vaak vanaf het derde levensdecennium. De been- en heupspieren worden aanvankelijk aangetast. Naarmate de ziekte voortschrijdt, breidt de spierzwakte zich ook uit naar de schouders en armen.
De manifestatie van het klinische beeld is vergelijkbaar met dat van juveniel SMA type 3. De progressieve spierzwakte is echter nog langzamer dan bij SMA type 3.
Wat veroorzaakt spinale musculaire atrofie?
Bij spinale musculaire atrofie gaan de tweede motorneuronen in het ruggenmerg verloren. Dit zijn zenuwcellen die met hun aanhangsels de spieren aansturen. Door de beschadiging van deze zeer gespecialiseerde motorneuronen bereiken er minder elektrische signalen de spieren dan bij gezonde mensen. Als spiercellen minder worden gebruikt en dus minder gestimuleerd worden, breekt het lichaam ze na verloop van tijd af.
Genetisch defect
In de meeste gevallen is spinale musculaire atrofie een erfelijke ziekte (erfelijke SMA). De oorzaak van de typische proximale SMA-vormen is onjuiste informatie in de genetische samenstelling van de patiënt. Het zogenaamde SMN1-gen op chromosoom 5 is niet functioneel.
Het SMN1-gen draagt de informatie - d.w.z. de blauwdruk - voor het vitale eiwitmolecuul genaamd SMN. SMN staat voor "Survival (of) Motor Neuron". Zonder het SMN-eiwitmolecuul gaan de motorneuronen na verloop van tijd verloren.
Het is waar dat er ook een verwant SMN2-gen in het lichaam is, dat in principe in staat is om de niet-functionele SMN1-genetische informatie te "compenseren". Maar dit gebeurt meestal maar in beperkte mate. Dit betekent dat een functieverlies van het SMN1-gen (indien onbehandeld) meestal niet volledig kan worden gecompenseerd door een intacte SMN2-genkopie.
Autosomaal recessieve en autosomaal dominante overerving
De genetische informatie van een persoon is in tweevoud beschikbaar. Als gevolg hiervan heeft iedereen twee exemplaren van het SMN1-gen - één van hun vader en één van hun moeder. De proximale spinale musculaire atrofieën van de kindertijd worden typisch overgeërfd als een autosomaal recessieve eigenschap.
Dit betekent dat beide genvarianten (allelen) van de ouders defect moeten zijn om spinale musculaire atrofie bij het nageslacht te laten ontwikkelen. Bij recessieve overerving worden de ouders niet aangetast omdat ze naast de functieloze ook een gezond SMN1-gen hebben dat het defect compenseert.
Ongeveer elke 45e persoon is de eigenaar van dit systeem voor SMA. Een koppel waarvan beide partners drager zijn, heeft 25% kans om een kind met de ziekte te krijgen.
In enkele gevallen in de adolescentie volgen vooral spinale musculaire atrofieën op volwassen leeftijd ook een autosomaal dominante overerving. In het geval van dominante overerving doet een defect gen zich al gelden - en worden de getroffenen ziek. Dit is echter niet het bovengenoemde genetische defect op chromosoom 5. Deze 5q-geassocieerde SMA's worden altijd autosomaal recessief overgeërfd.
Overerving met andere SMA-vormen
Niet-proximale spinale musculaire atrofie kan ook worden geërfd. De speciale spinobulbaire vorm (Kennedy-type) wordt recessief overgeërfd via een geslachtschromosoom, het X-chromosoom (dit heeft invloed op de genvarianten die de blauwdruk bevatten voor aanlegplaatsen voor mannelijke geslachtshormonen). Bij sporadische vormen is de overerving echter niet gegarandeerd. Er is hier weinig bekend waarom precies de tweede motorneuronen vergaan.
Onderzoeken en diagnose
De diagnose spinale musculaire atrofie wordt meestal gesteld door kinderartsen, kinderartsen die gespecialiseerd zijn in zenuwaandoeningen (neuropediatricians) en specialisten in ziekten van het zenuwstelsel (neurologen). Voor een meer precieze opheldering zijn verschillende onderzoeken nodig. Bij een SMA zijn vooral genetische tests en onderzoeken van zenuwen en spieren van belang.
Verzamelen van de medische geschiedenis (anamnese)
Bij elke ziekte vraagt de arts eerst naar de symptomen die zijn opgetreden en hoe het tot nu toe is verlopen. Bij baby's en peuters melden ouders veranderingen en afwijkingen in het gedrag van hun kind. Met name bij erfelijke ziekten richten artsen zich ook op de medische geschiedenis van de familie.
Lichamelijke onderzoeken
Kortom, een arts stelt afwijkingen in de motorische ontwikkeling vast door het kind fysiek te onderzoeken. Zo wordt getest of de kinderen zelfstandig hun hoofd rechtop kunnen houden, zitten of hun armen of benen zelfstandig kunnen bewegen (afhankelijk van hun leeftijd).
Aanvullende inspanningstesten worden uitgevoerd bij oudere kinderen en volwassenen met verdenking op spinale musculaire atrofie. De arts kijkt hoeveel kracht de betrokkene kan opbrengen en hoe lang hij die kan vasthouden. Hij onderzoekt ook het uithoudingsvermogen.
Bovendien test de arts de reflexen, die meestal verzwakt of gedoofd zijn, vooral in het geval van uitgesproken spinale musculaire atrofieën. Hiervoor tikt hij met een hamer op verschillende pezen, bijvoorbeeld op de hiel of onder de knie, en controleert de reactie.
Genetische studies
De meest betrouwbare detectiemethode voor (erfelijke) spinale musculaire atrofie is genetische analyse.Artsen zoeken naar bewijs van een veranderd (gemuteerd) SMN1-gen en het aantal bestaande SMN2-kopieën.
De algemene regel is om (erfelijke) SMA zo vroeg mogelijk te diagnosticeren en te behandelen. Afhankelijk van de vorm en de beschikbare behandeling kan de motorische ontwikkeling positief worden beïnvloed voordat de motorneuronen van het ruggenmerg onherstelbaar worden beschadigd.
Verdere onderzoeken bij SMA
Als een SMA wordt vermoed, meten artsen vaak de geleidingssnelheid van de zenuwen (elektroneurografie) en spieractiviteit (elektromyografie). Indien nodig onderzoeken ze ook de spieren met behulp van echografie (myosonografie) of magnetische resonantie beeldvorming (MRI).
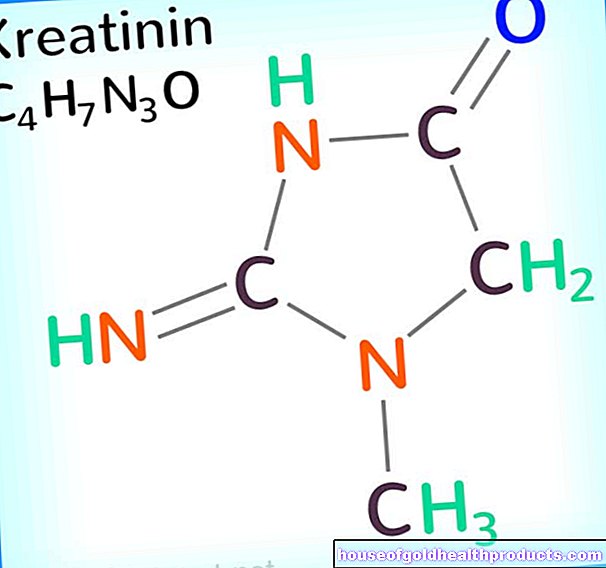
Daarnaast regelen artsen bloedonderzoeken. Als er spinale musculaire atrofie is, kunnen bepaalde parameters worden gewijzigd: bijvoorbeeld het niveau van creatinekinase (CK, een typisch spierenzym) wordt verhoogd.
Behandeling van spinale musculaire atrofieën
Behandeling van spinale musculaire atrofie is complex. Lange tijd was causale therapie voor geen enkele vorm van SMA mogelijk. Vooruitgang in medisch onderzoek biedt artsen echter nieuwe behandelingsopties om mensen met proximale SMA (SMN-gendefect op chromosoom 5) fundamenteel te helpen.
Bovendien concentreren artsen zich op het verlichten van de symptomen en het verlenen van de best mogelijke ondersteuning aan de getroffenen (bijvoorbeeld fysiotherapie, ademhalingstherapie, psychotherapie, mogelijk chirurgie).
Medische therapie
De nieuwe behandelmethoden voor patiënten bij wie de SMA is gebaseerd op een bekend SMN-gendefect grijpen rechtstreeks in in het genetisch materiaal zelf of in de downstreamverwerking van de genetische informatie.
Het doel is om het lichaam van de patiënt in staat te stellen zelfstandig voldoende hoeveelheden van het SMN-eiwit aan te maken, dat cruciaal is voor de motorneuronen.
De volgende behandelingsopties zijn beschikbaar voor spinale musculaire atrofie:
- Splitsingsmodulatoren (Nusinersen, Risdiplam): Deze geneesmiddelen grijpen direct in bij de verdere verwerking van boodschapper-RNA-moleculen. Ze versterken die processen die een grotere hoeveelheid SMN-eiwit afleveren uit het intacte SMN2-gen.
- Genvervangende therapie (Onasemnogene Abeparvovec): Deze therapie grijpt direct in in het menselijk genoom. De defecte kopie van het SMN1-gen wordt in de aangetaste cellen vervangen door een extern geleverd, functioneel genconstruct.
Splitsingsmodulatoren
In het geval van een SMN1-gendefect kan het lichaam ook het SMN-eiwit produceren uit het verwante SMN2-gen. Het vervangende gen SMN2 “springt erin”, maar dat is niet genoeg. De reden: De eiwitten in SMN2 zijn meestal te kort en worden snel afgebroken.
Dit komt door de verwerking van het overeenkomstige SMN2-boodschapper-RNA (SMN2-mRNA). Het verzendt de constructie-informatie van het genoom (DNA) naar de eiwitproductieplaatsen (ribosomen).
Hiervoor wordt eerst het SMN2-gen in het genoom uitgelezen. Een voorlopig SMN2-boodschapper-RNA wordt geproduceerd. Het moet onder andere verder verwerkt worden door middel van zogenaamde splicing. Pas dan ontstaat het rijpe boodschapper-RNA. Speciale celcomplexen, de ribosomen, lezen dan het rijpe boodschapper-RNA af en produceren zo het SMN2-eiwit. En juist dit is verkort en onstabiel, snel ontmanteld en kan de functie van SMN1 niet overnemen.
Om hierin verandering te brengen, beïnvloeden de actieve ingrediënten Nusinersen en Risdiplam de verdere verwerking van het voorlopige boodschapper-RNA. Hierdoor verhogen deze zogenaamde splicing-modulators uiteindelijk de hoeveelheid bruikbare SMN-eiwitten - en kunnen ze dus voor een adequate toevoer zorgen.
Nusinersen
Het medicijn Nusinersen is een zogenaamd "antisense-oligonucleotide" (ASO). Het werd in 2017 goedgekeurd door het Europees Geneesmiddelenbureau. ASO zijn kunstmatig geproduceerde en speciaal aangepaste RNA-moleculen. Ze binden specifiek en precies aan het SMN2-boodschapper-RNA. Dit voorkomt dat ze in de menselijke cel verkeerd verder worden verwerkt.
Concreet: Nusinersen voorkomt dat belangrijke informatie (exon 7) per ongeluk uit het SMN2-boodschapper-RNA wordt geknipt. De verblijfplaats van exon 7 zorgt ervoor dat het lichaam vervolgens meer functioneel SMN-eiwit maakt.
Nusinersen wordt toegediend via een zogenaamde lumbaalpunctie. Dit betekent dat het medicijn met een injectiespuit in het wervelkanaal wordt geïnjecteerd. Deze therapie wordt herhaald met regelmatige tussenpozen van enkele maanden. In het eerste jaar van de behandeling krijgen de getroffenen zes en vervolgens drie doses per jaar.
Patiënten verdragen het medicijn meestal goed. Nusinersen leidt tot gunstiger ziekteverloop. Studies hebben aangetoond dat de mobiliteit bij veel patiënten verbetert: in veel gevallen was het mogelijk om vrij te zitten en het lichaam zelfstandig te draaien. Bijwerkingen en complicaties zijn onder meer gebaseerd op de lumbaalpunctie (bijvoorbeeld hoofdpijn, hersenvliesontsteking).
Risdiplam
De Europese Commissie keurde Risdiplam goed als het derde medicijn tegen 5q-geassocieerde SMA (types 1-3 of één tot vier SMN2-genkopieën) in maart 2021. Risdiplam wordt dagelijks ingenomen als een opgelost poeder. De exacte dosis wordt berekend op basis van leeftijd en lichaamsgewicht.
In tegenstelling tot Nusinersen is Risdiplam geen "antisense-oligonucleotide", maar een klein molecuul. Dit molecuul bindt aan het boodschapper-RNA voor SMN2-eiwitten en stabiliseert ze op deze manier. Hierdoor ontstaan meer functionele SMN-eiwitten.
Vaak voorkomende bijwerkingen van Risdiplam zijn gastro-intestinaal ongemak, huiduitslag, koorts en urineweginfecties.
Genvervangende therapie
Een andere benadering van de behandeling van proximale spinale musculaire atrofie is gebaseerd op wat bekend staat als genvervangingstherapie. Het defecte SMN1-gen - het startpunt van de progressieve SMA - wordt "vervangen" door een nieuwe functionele kopie van het gen.
De werkzame stof Onasemnogene Abeparvovec (AVXS-101), die volgens dit principe werkt, kreeg in mei 2020 een voorwaardelijke handelsvergunning voor de behandeling van peuters en kinderen van het Europees Geneesmiddelenbureau (EMA).
Het medicijn kan worden gebruikt voor SMA type 1 volgens EMA-informatie. Bij alle andere vormen van SMA-ziekte bepalen genetische kenmerken (aantal SMN2-kopieën) of genvervangingstherapie een optie is.
Met Onasemnogene Abeparvovec wordt een functionele kopie van het menselijke SMN1-gen ingebracht in de aangetaste cellen van het ruggenmerg en de hersenstam. Dit wordt gedaan door bepaalde virussen die dienen als "veerboten" voor het nieuwe genetische materiaal - zogenaamde adeno-geassocieerde virale vectoren (AAV-vectoren).
De vectorgenconstructen worden eenmalig als een infuus via de ader in de bloedbaan gegeven en van daaruit door het lichaam verspreid. Door een nog niet volledig ontwikkelde bloed-hersenbarrière bij kleine kinderen kunnen deze vectoren ook in het ruggenmergweefsel terechtkomen.
Door deze vectoren bij voorkeur te binden aan speciale oppervlaktestructuren van de motorneuronen, nemen ze bij voorkeur het genetische materiaal op om vervolgens zelfstandig het SMN-eiwit te produceren.
Behandeling kan motorische functies verbeteren en leiden tot blijvend ontwikkelingssucces (bijvoorbeeld zitten, kruipen en lopen zonder ondersteuning).Tijdens de behandeling kunnen de leverwaarden soms flink oplopen, maar kan het aantal bloedplaatjes afnemen. Koorts en braken komen ook vaak voor. Om bijwerkingen te verminderen, krijgen patiënten gedurende enkele weken corticosteroïden ("cortison").
Leeftijdsgebonden motorische ontwikkeling is over het algemeen alleen mogelijk als gentherapie presymptomatisch is gestart. De behandeling vindt plaats in gespecialiseerde neuromusculaire behandelcentra.
fysiotherapie
Fysiotherapie blijft een belangrijke pijler van de behandeling van SMA Niet elke vorm van SMA kan met de nieuwe behandelmethoden worden behandeld. Regelmatige oefentherapie is bedoeld om de fysieke capaciteiten op peil te houden en de spierafbraak te vertragen.
De fysiotherapeut beweegt passief lichaamsdelen die al verlamd zijn. Actieve bewegingssequenties worden op hun beurt getraind om de mobiliteit en kracht van de spieren te ondersteunen. Massage of warmte- en koudebehandelingen kunnen ook helpen. Deze dienen ook om te ontspannen en, onder bepaalde omstandigheden, verdere degeneratie te vertragen.
Logopedie
In sommige gevallen beïnvloedt SMA de spreek- en slikspieren. Dan helpt een logopedische oefening. Het stimuleert kinderen om te leren spreken. Zelfs bij oudere patiënten kan dit gewoonlijk een verslechtering van de spraak vertragen. Logopedisten trainen ook correct slikken.
Zowel fysiotherapeuten als logopedisten ondersteunen de getroffenen met gerichte ademhalingstherapie.
Pijnstillende behandeling
Pijntherapie speelt een belangrijke rol, vooral in de meer gevorderde stadia van de ziekte. Artsen gebruiken pijnstillers om het lijden van de getroffenen te verminderen.
chirurgie
Omdat spinale musculaire atrofie kan leiden tot ernstige kromming van de wervelkolom (scoliose), overwegen artsen soms een operatie. Daarbij verstevigen ze specifiek de wervelkolom.
Dit geeft de getroffenen een (zekere) extra rompstabiliteit, wat niet alleen een meer rechtopstaande houding mogelijk maakt, maar ook botten en gewrichten beschermt. Een wervelkolomoperatie kan ook helpen tegen progressieve ademhalingsproblemen.
Psychotherapeutische zorg
Neuromusculaire ziekten zoals spinale musculaire atrofie vertegenwoordigen grote psychologische stress.Patiënten en familieleden verwerken de diagnose in individuele en groepssessies onder leiding van psychotherapie en ontwikkelen strategieën om beter met de ziekte om te gaan.
Ook zelfhulpgroepen en patiëntenvertegenwoordigers bieden belangrijke ondersteuning. Ze informeren, adviseren en ondersteunen de getroffenen en hun familieleden om de uitdagingen van een SMA-ziekte het hoofd te bieden.
Kans op herstel van spinale musculaire atrofieën
Als er spinale musculaire atrofie is, hangt de prognose voornamelijk af van de respectieve vorm. Hoe later de symptomen verschijnen, hoe beter het beloop. Bovendien, hoe eerder artsen spinale musculaire atrofie diagnosticeren, hoe eerder ze geschikte behandelingsmaatregelen kunnen starten, zelfs voordat motorneuronen onomkeerbaar zijn beschadigd.
De nieuwe behandelingsopties door middel van splicing-modulatoren en genvervangingstherapie hebben een groot potentieel bij de behandeling van proximale SMA - vooral wanneer de behandeling (zeer) vroeg wordt gestart. Gegevens voor een betrouwbare langetermijnprognose zijn echter nog in behandeling. Alleen verdere studies en nauwgezette observaties van de veiligheid van geneesmiddelen kunnen de komende (maanden en) jaren meer zekerheid bieden. Met de nieuwere medicijnen is langdurige beheersing van de ziekte of zelfs genezing op zijn minst denkbaar.
SMA type 1 is over het algemeen een ernstige ziekte Kinderen die SMA type 1 krijgen, hebben (onbehandeld) een zeer beperkte levensverwachting. De snel toenemende spierzwakte over het hele lichaam beïnvloedt ook de ademhaling. De gevolgen zijn acute longontsteking en zelfs ademhalingsfalen. Getroffen kinderen overlijden binnen de eerste levensjaren.
De prognose is iets beter voor SMA type 2. De levensverwachting varieert afhankelijk van de exacte ernst van de ziekte: sommigen sterven in de kindertijd, maar de meeste bereiken de jonge volwassenheid. Vroeg of laat moet - indien gewenst - de ademhaling in de meer ernstige vormen worden ondersteund. Getroffen mensen blijven mobiel met behulp van een rolstoel.
Met SMA type 3 is de prognose aanzienlijk beter - vooral als de eerste symptomen laat verschijnen. De prestaties verslechteren geleidelijk in de loop van een aantal jaren. Op oudere leeftijd kan een rolstoel of zelfs permanente zorg nodig zijn. De levensverwachting wordt nauwelijks beperkt door type 3 spinale musculaire atrofie.
Spinale musculaire atrofie bij volwassenen (type 4) is zelfs langzamer dan type 3. Mensen hebben meestal een normale levensverwachting.
Tags: sport fitness Ziekten nieuws